
Table of Contents
- Chemsmart is an open-source “Swiss Army knife” toolkit designed to automate the tedious manual processes in computational chemistry, letting scientists focus on scientific thinking instead of file format conversions.
- A new framework gives AI generative models a “physics tutor.” It uses reinforcement learning to reward chemically stable and realistic molecules, guiding AI beyond simple imitation.
- DrugReasoner is an AI that predicts the probability of a new drug’s approval. It explains its predictions by comparing known successful and failed drugs, reasoning like a chemist and revealing the model’s internal logic.
- A new process uses known molecular fragment data to guide and correct traditional docking. This improves the accuracy of predicting how a drug molecule binds, showing the power of combining data with physics.
- DRUGFLOW learns the complete distribution of “good molecules,” like drawing a complete treasure map. This allows for more realistic and controllable molecule generation.
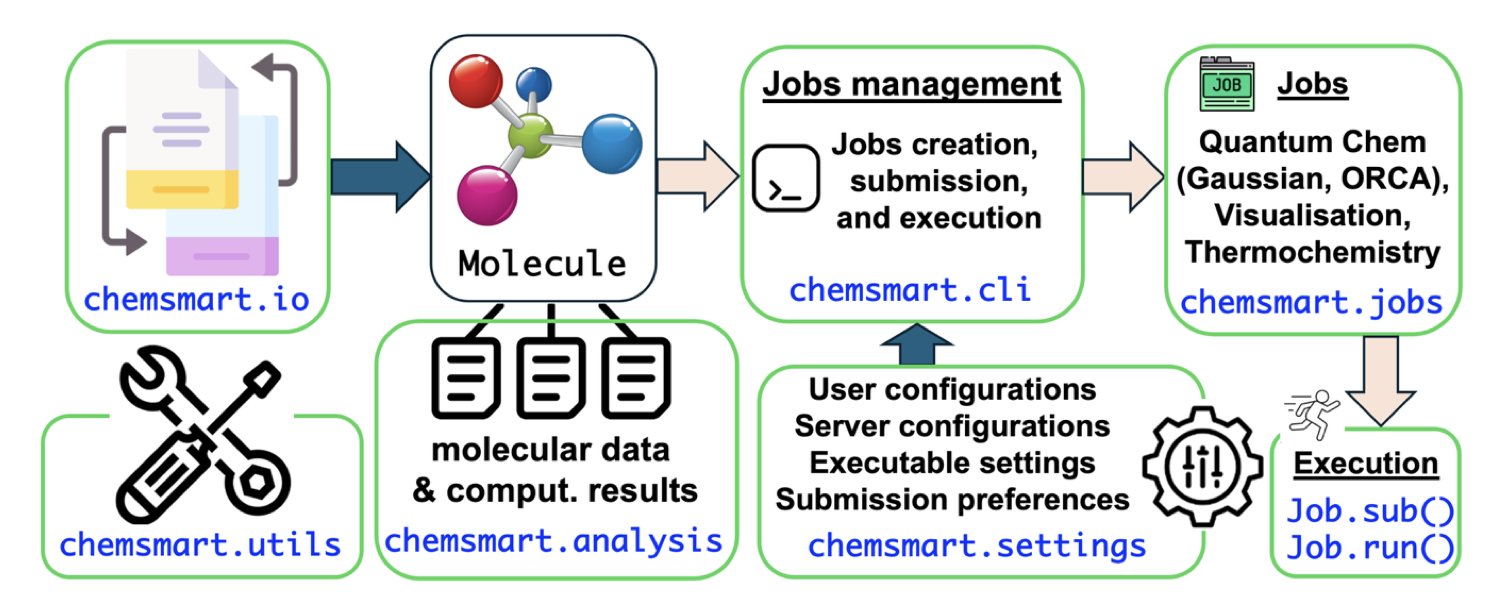
1. Chemsmart: Saying Goodbye to the ‘Manual Workshop’ of Computational Chemistry
Anyone who’s done quantum chemistry calculations by hand knows the deal: most of your time is spent wrestling with tedious data, like a data janitor. Very little time is left for thinking about the deep chemistry questions.
A researcher’s typical workflow looks like this: you draw a molecule in one program and save it in a specific format. Then you convert it to another format for the calculation. Next, you have to manually write an input file, carefully set the parameters, and submit it to a computing cluster. While you wait, you constantly check the job status. When it’s done, you have to extract key data like energy and coordinates from a massive output file, either by hand or with a custom script. Finally, you import this data into visualization software to make images for your report.
Every step in this process is filled with repetitive work, format incompatibilities, and the risk of human error. We have powerful computational engines like Gaussian and ORCA, but we’re still stuck manually “refueling and cleaning them.”
Chemsmart: A “Project Manager” for Computational Chemistry
The Chemsmart toolkit aims to be a “project manager” for the entire research process, solving the real pain points in research.
As an open-source Python toolkit, Chemsmart’s idea is simple: automate the most tedious manual tasks in computational chemistry.
Here’s how it works:
First, it defines a core Molecule object, which you can think of as a “universal file format.” Molecules and data from different chemistry software can all be converted into this standard object. This solves the format incompatibility problem, like giving all software a universal translator.
Second, it breaks down the entire computational process into independent, modular tasks, like “geometry optimization,” “transition state search,” and “thermodynamic analysis.” Users can combine these modules like Lego bricks to build fully automated workflows.
A user just needs to give a command, like: “Optimize this molecule, calculate its frequencies, and generate a PyMOL visualization file.” It handles the rest. Chemsmart automatically prepares input files, submits jobs, monitors progress, parses the results, and organizes all the files.
The Change Chemsmart Brings
Researchers are freed from repetitive, uncreative labor and can focus on the chemistry itself.
For beginners, Chemsmart lowers the entry barrier. Users don’t need to memorize the obscure input parameters for various software; they just call a few simple Python functions.
For experienced researchers, it boosts efficiency and reproducibility. It becomes possible to run standardized computational workflows on hundreds of molecules, which is critical for studies like structure-activity relationships.
All of Chemsmart’s code is open source and has a modular design. This means it can be used as a standalone tool or as a Python library, integrating seamlessly into more complex, custom analysis pipelines.
Chemsmart is a practical toolbox built by an experienced engineer to solve everyday problems. For researchers on the front lines, a handy toolbox is sometimes more valuable than a distant theory.
📜Title: Chemsmart: Chemistry Simulation and Modeling Automation Toolkit for High-Efficiency Computational Chemistry Workflows 📜Paper: https://arxiv.org/abs/2508.20042v1
2. AI Molecule Generation: Finally Learning Physics
Anyone who has worked with 3D molecule generation models knows the reality: these AIs are first-rate artists but last-rate engineers.
You give one a task, and it can “draw” molecules that are structurally novel and look beautiful. But when you put these “artworks” under a physics microscope, problems appear. Bond lengths are wrong, bond angles are distorted, and atoms are packed together like a rush-hour subway car. These molecules couldn’t exist in the real world.
We’ve always wanted an AI engineer that understands physics. The new RLPF framework is a solid step in that direction.
Getting AI a Physics Tutor
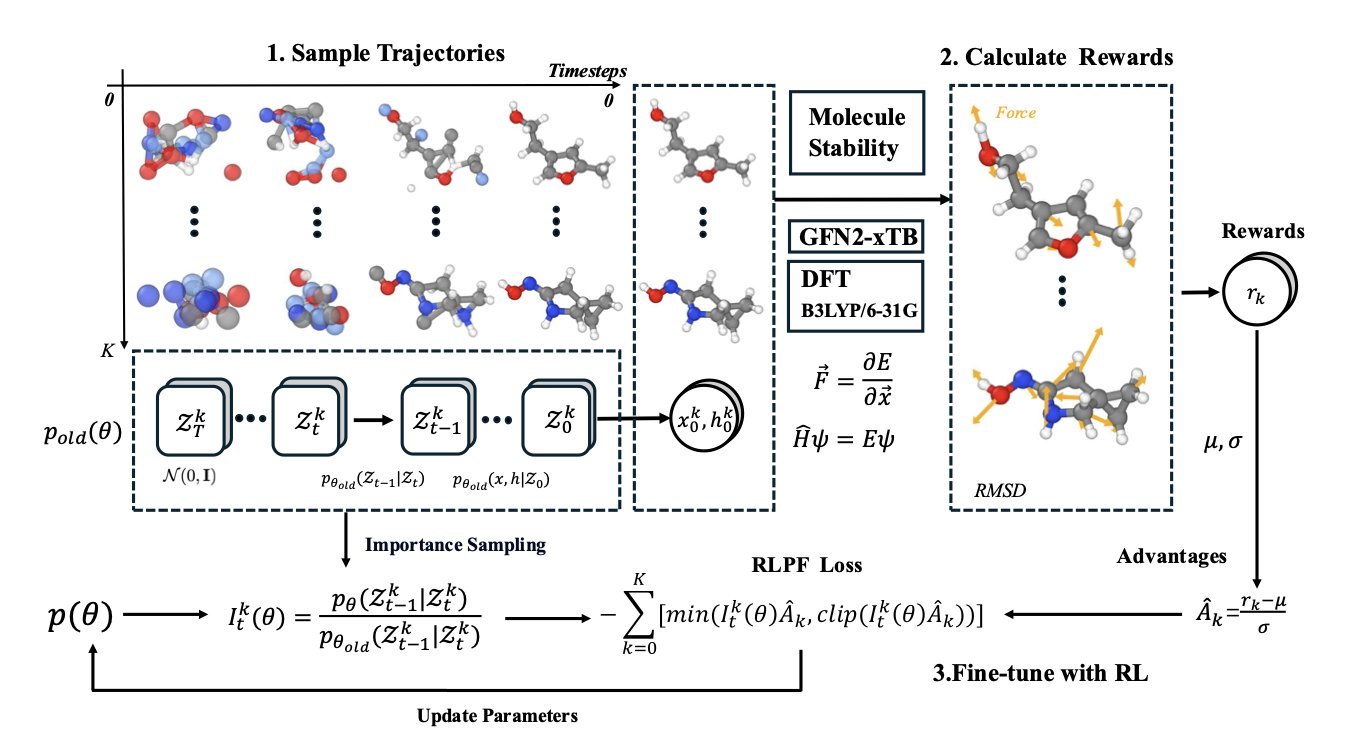
The researchers didn’t build a more complex AI that had seen more data. Instead, they took a different path. They gave the AI a “physics tutor” and a “reward-and-punishment” system.
The tutor is a force field, and the system is Reinforcement Learning (RL).
Here’s how it works:
A diffusion model doesn’t generate a molecule all at once. It’s like a sculptor starting with a fuzzy “cloud of atoms” and gradually carving out the details.
The RLPF framework has the physics “tutor” evaluate the sculpture after every single cut.
The tutor (the force field) determines if that cut increased or decreased the internal stress of the structure. Did it make it more stable or closer to falling apart?
If a step makes the molecule more stable and physically sound, the AI gets a “reward” and remembers it was a good move.
If a step makes the molecule more unstable, the AI gets a “punishment” and learns to avoid that path.
Through this constant loop of physics feedback, rewards, and punishments, the AI is forced to consider the physical consequences of every creative step. It moves beyond just imitating the shapes of molecules in its training data.
How Well Does It Work?
The molecules generated by the tutored AI are on another level.
On the standard QM9 dataset, the proportion of physically stable molecules it generated rose from 82% to over 93%.
This “tutor” framework is versatile and can guide different diffusion models like EDM, GeoLDM, and UniGEM. It’s a universal, plug-and-play “enhancement module.”
This work steps outside the paradigm of competing on model parameters and represents a fundamental shift in thinking. It injects physics knowledge directly into the AI’s generation process.
📜Title: Guiding Diffusion Models with Reinforcement Learning for Stable Molecule Generation 📜Paper: https://arxiv.org/abs/2508.16521 💻Code: https://github.com/ZhijianZhou/RLPF/tree/verl_diffusion
3. DrugReasoner: An AI That Can Explain Its Predictions
The field of drug discovery is full of AI models that can predict a molecule’s solubility, toxicity, or binding affinity. But very few people would dare to bet a hundred-million-dollar clinical trial entirely on a number from an AI model.
That’s because most of these models are black boxes.
When a model gives a druggability score of 0.87 but can’t explain why, it’s like a fortune teller who only gives you numbers. Decision-makers can’t tell if the prediction is based on rigorous calculation or a random guess. A result that can’t be explained has limited value.
DrugReasoner was designed to solve this problem.
The AI Learns to “Reason by Analogy”
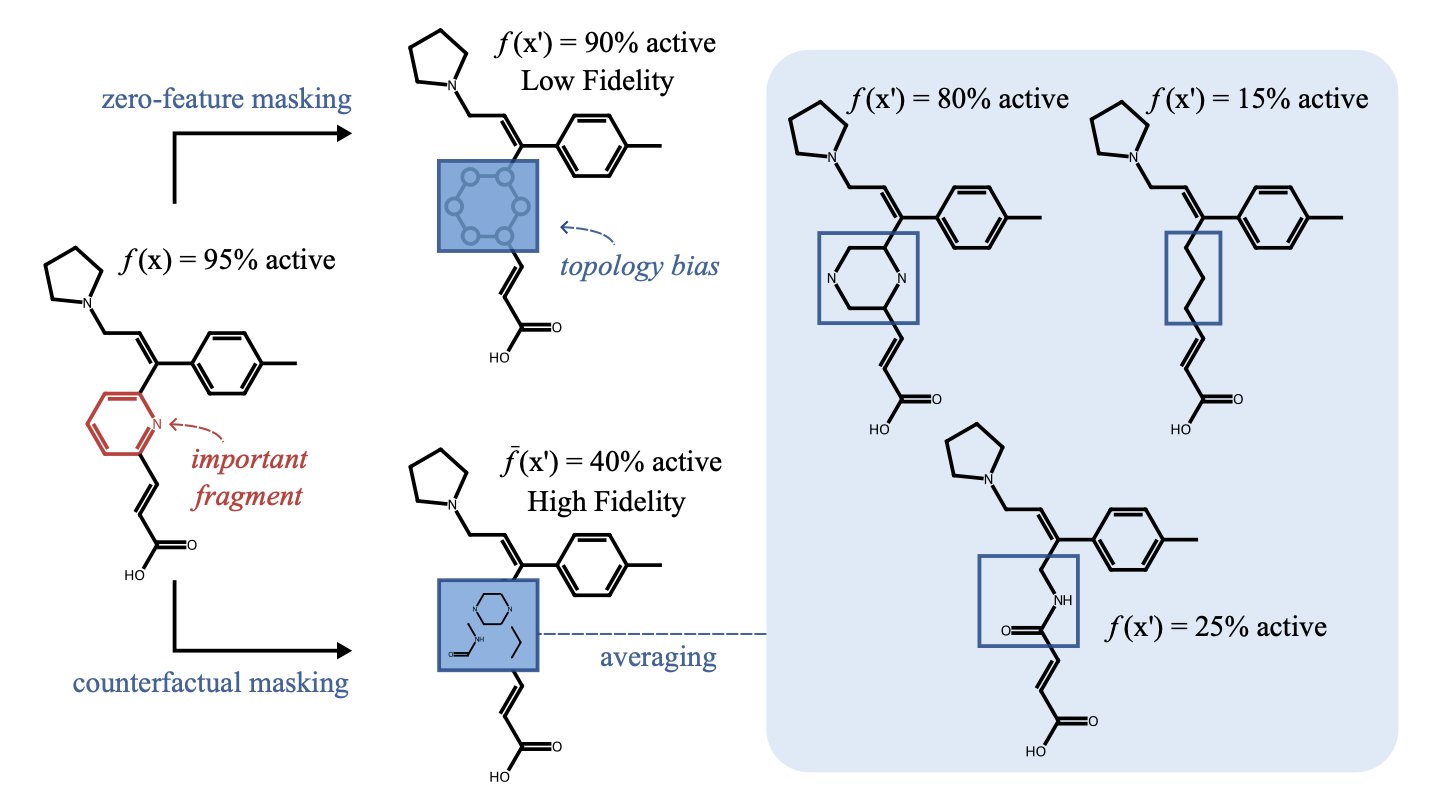
The researchers trained the AI to master a core skill of chemists: comparison.
When you input a newly designed drug candidate, DrugReasoner searches its knowledge base to find the most structurally similar known drugs. It then divides these similar drugs into two groups: those that made it to market successfully and those that failed in clinical trials.
Finally, it generates a reasoning report, for example:
“This molecule has a 73% probability of approval. Part A of its structure is similar to a successful anti-cancer drug, and this structure generally has good druggability. However, Part B is similar to a molecule that failed due to liver toxicity, which lowers its probability of approval.”
This type of explanation provides clear information that can be used for decision-making.
What Were the Results?
When tested on a dataset containing a large number of real drug molecules, DrugReasoner’s prediction accuracy (AUC of 0.73) was comparable to traditional machine learning methods like XGBoost and better than another recently published model, ChemAP.
This shows that the model provides explainability without sacrificing predictive accuracy.
Predicting whether a new drug will be approved depends on many variables, including science, business, and luck. In this context, an AUC of 0.73 is a solid result, though not perfect.
The researchers acknowledge that to prevent the model from “cheating” by memorizing SMILES strings, the SMILES representations of the molecules were not provided during training. This might have limited the model’s understanding of fine-grained chemical structures, but it also proves that the reasoning method is effective even with limited information.
📜Title: DrugReasoner: Interpretable Drug Approval Prediction with a Reasoning-augmented Language Model 📜Paper: https://arxiv.org/abs/2508.18579 💻Code: https://github.com/mohammad-gh009/DrugReasoner
4. Polaris Challenge: Using Fragment Knowledge to Improve Docking Accuracy
Researchers in structure-based drug design have a complicated relationship with molecular docking software. It’s fast, capable of screening a million-compound library overnight. But its scoring functions are often unreliable. The software will generate lots of seemingly perfect binding poses and give them high scores, but experimental validation often proves them wrong.
Docking software is like a fast but reckless scout. It can survey the entire battlefield quickly but often mistakes scarecrows for the enemy. What researchers need is a pair of “friend-or-foe” goggles. A paper from the Polaris Challenge demonstrates just such a pair.
Stop Letting AI Guess, Give It a “Cheat Sheet”
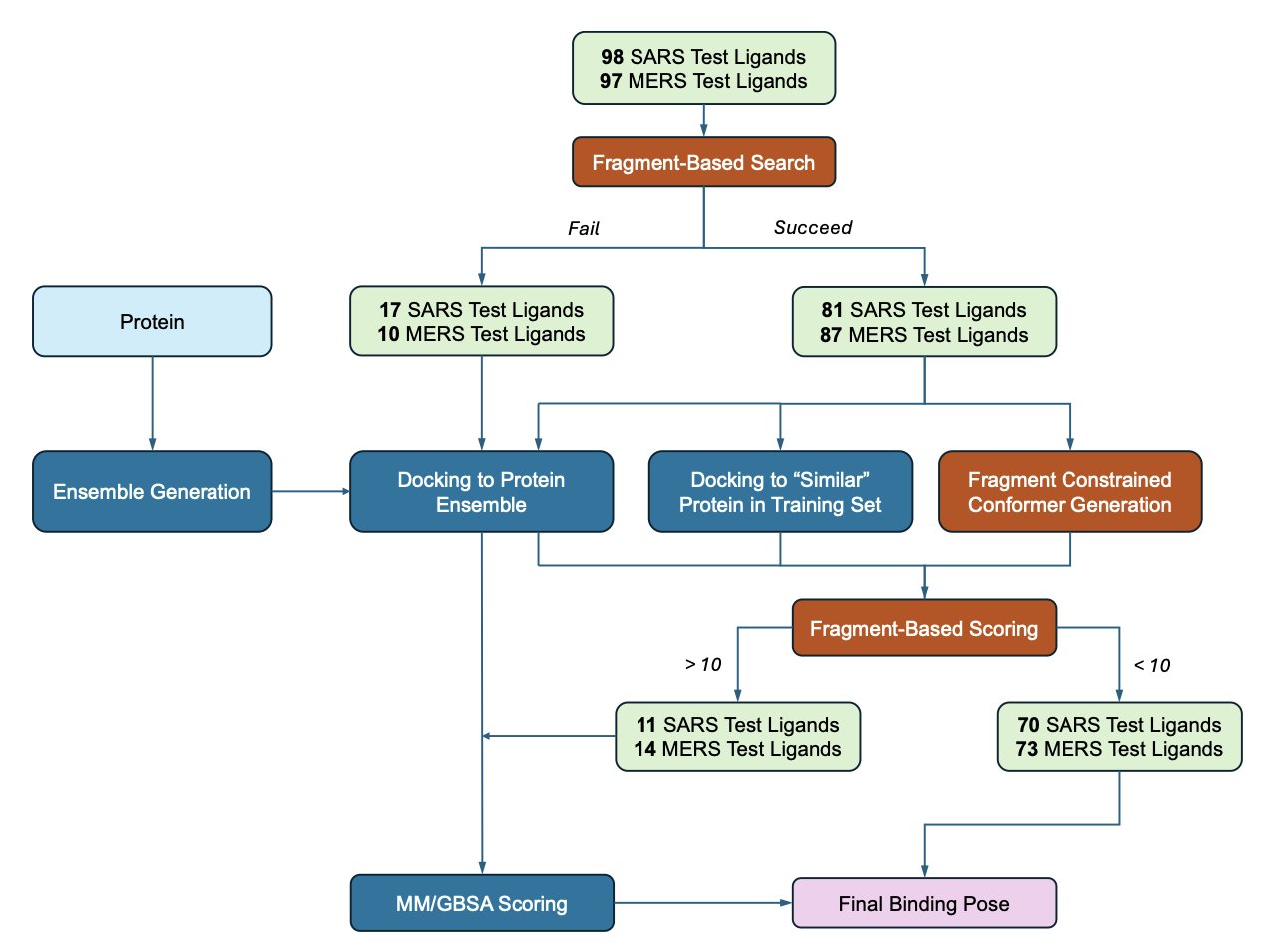
The researchers’ idea wasn’t to rebuild the docking engine but to improve the scoring step. They argue that tools like Vina are already good enough at generating diverse poses; the weak link is the scoring.
The “cheat sheet” they used is the crystal structures of thousands of small molecule fragments from a database.
These fragments act like “road signs,” using experimental data to mark which areas of a protein pocket are hydrophobic and where hydrogen bonds are likely to form.
The new process first uses VinaGPU to generate many possible binding poses. Then, the AI uses the “cheat sheet” to grade them. It checks whether the chemical groups in the molecule match the “road signs” from the known fragments. A match gets bonus points; a mismatch gets points deducted.
The process also uses the “cheat sheet” more proactively through constrained docking. Like a hint before an exam, it guides the docking program to search in the right direction from the very beginning.
What If There’s No Cheat Sheet?
When there’s no relevant fragment data to reference, the process switches to a backup plan: MM/GBSA rescoring.
This traditional physics-based calculation method is slower than Vina’s scoring function but is generally more accurate.
The entire process forms a multi-layered filter: screen with the fastest method, refine with data-driven knowledge, and finally, double-check with physics.
Does This Thing Actually Work?
The researchers applied this process to the proteases of the COVID-19 virus (SARS-CoV-2) and its close relative, MERS-CoV.
The results showed that for over half of the molecules, the process successfully predicted a binding pose with an error of less than 2 angstroms compared to the crystal structure. This is a solid performance on a real-world target.
They also demonstrated that the knowledge learned from SARS-CoV-2 fragment data could be successfully transferred and used for predictions on MERS-CoV.
This work offers a deployable engineering solution designed to solve practical problems in day-to-day research. The future of computational drug discovery lies in cleverly combining data, physics principles, and chemical intuition, not in relying on a single tool.
📜Title: Polaris Challenge: Data-driven Priors to Improve Docking for Pose Prediction 📜Paper: https://doi.org/10.26434/chemrxiv-2025-fv3c3
5. DRUGFLOW: AI Learns to Draw a “Treasure Map”
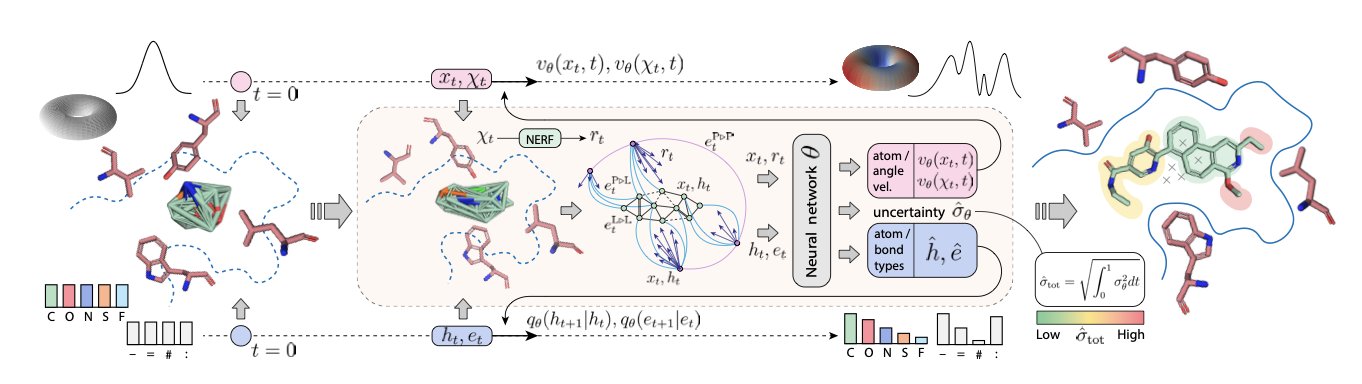
- It learns the entire distribution of “good molecules,” like drawing a complete treasure map, instead of the old method of just marking individual treasure spots.
- It has built-in features chemists need, like uncertainty detection and protein flexibility, turning the AI from an artist into an engineer.
Anyone who has worked with generative AI knows that it’s a first-rate artist but a second-rate chemist. Give it a protein pocket, and it can “draw” novel molecules that look perfect. But most of these molecules just statistically “resemble” known drugs. The AI doesn’t really understand what makes a molecule “good.”
It’s like an artist who can only copy Van Gogh’s paintings. He can paint a starry night, but he doesn’t understand its deeper meaning.
DRUGFLOW’s goal is to teach the AI to learn astronomy.
From “Taking Snapshots” to “Drawing a Map”
The core idea of this work is a shift in perspective. A good drug sits within a vast “chemical space” in a region of low energy and good properties. The AI’s job, then, is to learn to draw a complete “map” of this region, rather than just generating single “snapshots.”
DRUGFLOW learns the complete distribution of all “good molecules” in a protein pocket by combining two mathematical tools: Continuous Flow Matching, which you can think of as tracing the terrain with smooth curves, and Discrete Markov Bridges, which you can imagine as building step-by-step paths between key landmarks.
Once it learns to draw the map, the AI transforms from an imitating artist into a geographer who can point out the peaks and valleys.
The AI Finally Grows a “Brain”
With this global view, the AI can now perform functions that were difficult before.
First is uncertainty. DRUGFLOW has a built-in “unreliability detector.” When it generates a molecule that falls in an unexplored region of the map, it will tell you directly: “I’m not sure if this molecule is reliable.” This ability to know what it “doesn’t know” can prevent scientists from synthesizing molecules that the AI just hallucinated.
Second is size adaptation. When generating a molecule, it dynamically adjusts its size, proactively removing atoms that might clash with the protein pocket. It’s like a mover who takes the legs off a sofa before trying to fit it through a doorway.
Finally, there’s protein flexibility. An upgraded version, FLEXFLOW, can adjust the orientation of amino acid side chains in the protein pocket while simultaneously generating the ligand molecule. In the real world, a protein pocket isn’t a rigid mold; it’s more like a soft glove. FLEXFLOW acts like a true designer, allowing the glove to adapt to the shape of the hand as the hand is being designed.
📜Title: Multi-Domain Distribution Learning for De Novo Drug Design 📜Paper: https://arxiv.org/abs/2508.17815