
Table of Contents
- A new machine learning method can quickly and accurately predict hydrogen bond strength with 3% error by focusing on a few key quantum chemistry features, and it runs on a standard laptop.
- Researchers are building a “virtual human” driven by a team of AI experts to simulate a drug’s complete journey, from molecule to whole-body effects, tackling the biggest gap in drug development.
- MolPrompt gives AI a chemistry “cheat sheet” (a knowledge prompt), forcing it to learn a molecule’s physicochemical properties along with its graph, helping it gain deeper, more useful chemical knowledge.
1. AI Predicts Hydrogen Bond Strength: Fast, Accurate, and Cheap

Hydrogen bonds are everywhere in chemistry and biology. They hold together the DNA double helix, determine how proteins fold, and control how drug molecules bind to their targets. But measuring the strength of a hydrogen bond isn’t easy.
In the past, you had two choices: guess using simple rules of thumb, which is fast but inaccurate, or run high-precision quantum chemistry calculations on a supercomputer, which is accurate but takes days and costs a lot. For a long time, the field has needed a tool that offers both speed and accuracy. A recent study provides a solution.
The AI Learned to Ask the Right Questions
The idea behind this work was not to build a black-box model that handles the entire complex molecular system. Instead, it was to guide the AI to focus on a few fundamental physical quantities.
Here’s how the model works. The researchers first run a quick calculation on the system using standard quantum chemistry programs. But instead of using the full 3D structure, they extract a few key values:
- The partial charge of the hydrogen bond acceptor atom.
- The partial charge of the hydrogen bond donor atom.
- The bond order between the hydrogen atom and its covalently bonded donor atom in the hydrogen bond.
This process is like predicting a car’s speed. Instead of showing the AI a photo of the whole car, you give it core parameters like engine displacement, vehicle weight, and tire size. The AI’s task shifts from complex image recognition to a simpler regression problem that gets closer to the heart of the matter.
When the AI receives these “gold-standard” features, which hold physical and chemical meaning, its learning efficiency and accuracy both improve.
How Were the Results? Fast and Good
The researchers fed these features into a model that combines two machine learning methods: Support Vector Regression and gradient boosting. The model’s mean absolute percentage error was just 3%. For hydrogen bond energy, a subtle interaction easily affected by its environment, this level of precision is good enough to guide most drug design and materials science research.
The model can also handle “cooperative effects” that are difficult for simple linear models. In real systems, hydrogen bonds don’t exist in isolation; they form networks and influence each other. The new model can capture these complex, non-linear relationships.
Another key advantage of this work is efficiency.
The researchers found that calculating the input features didn’t require the most expensive and precise quantum chemistry methods. A more affordable density functional method (BLYP) and a more expensive hybrid density functional method (B3LYP) produced nearly identical final predictions.
They also tested an even cheaper semi-empirical method (xTB2). The results showed that while accuracy dropped slightly, it was still suitable for quick preliminary analysis.
Users no longer need a computing cluster. They can get reliable estimates of hydrogen bond strength for their systems quickly on a standard laptop.
This method is a practical tool with broad applications, not just a theoretical exploration. It has the ability to change how daily work is done in related research fields.
📜Title: Estimating the Hydrogen Bond Strength by Machine Learning Approaches 📜Paper: https://chemrxiv.org/engage/chemrxiv/article-details/68a47621728bf9025e2d81c4 💻Code: 10.5281/zenodo.16902603
2. The AI Virtual Human: Drug Discovery’s Ultimate Flight Simulator
The biggest pain point in drug development is the translation gap.
A compound can show great results in cell and animal experiments, only to fail when it enters human clinical trials. Countless projects and a lot of money have been lost this way. Existing computational models and AI tools are too fragmented. Some are good at molecular docking, others at predicting pharmacokinetics, but they all work in isolation and can’t see the big picture.
This paper envisions a framework that can integrate the big picture: building a “virtual human” in a computer.
This Isn’t One AI, It’s a Team
The feature of this Full-Body AI-Agent framework is that it doesn’t build a single, all-powerful AI. Instead, it assembles a team that works together.
The framework is like a biomedical R&D company with seven core departments:
Each department is a specialized AI agent with its own expert data and models. A central coordinating Large Language Model (LLM) acts as the “project manager” for the entire team.
How Does the AI Hold a Project Meeting?
The framework operates like a project meeting.
For example, to assess the long-term liver toxicity of a new drug, the “project manager” LLM breaks down the task. It first directs the “molecular department” AI to analyze how the drug interacts with various enzymes in liver cells. Then, it passes the molecular-level results to the “cell department” AI to simulate the liver cells’ response. Next, the “tissue” and “organ” AIs use the cellular changes to predict the impact on the entire liver’s structure and function.
This process is a two-way street. If the “organ department” AI detects abnormal liver function indicators, it sends that information back to the “project manager.” The project manager might then go back to the “molecular department” AI to see if the problem comes from a specific chemical group on the drug and if the molecular structure can be changed to avoid it.
This kind of cross-level, iterative communication simulates a real research process.
Two Practical Drills
To show this is feasible, the authors designed two use cases.
The first is a tumor metastasis agent. It simulates the process of cancer metastasis: how a tumor cell detaches from its original site, enters the bloodstream, and settles in another organ. This AI team would analyze molecular signals, cell behavior, and the whole-body physiological environment to produce a “metastasis risk score.”
The second is a drug agent. It guides preclinical evaluation in a computer, like testing a drug on an organ-on-a-chip. It doesn’t just analyze the chip data; it also considers the drug’s distribution and metabolism in other organs to provide a prediction closer to what would happen in a human body.
Creating a complete “digital twin” of a person is still a long way off. This framework requires a huge amount of high-quality, standardized, multi-dimensional data. But it lays out a blueprint.
The future of AI in drug development may not depend on a single algorithmic breakthrough. It might depend on how we organize knowledge and models at different scales, much like managing an efficient team. This framework won’t replace scientists, but it can provide an unprecedented “flight simulator,” allowing us to see which route is safest for a drug and where storms might lie before it ever enters a human body.
📜Paper: https://arxiv.org/abs/2508.19800
3. MolPrompt: Teaching AI to Understand Molecules with Knowledge Prompts
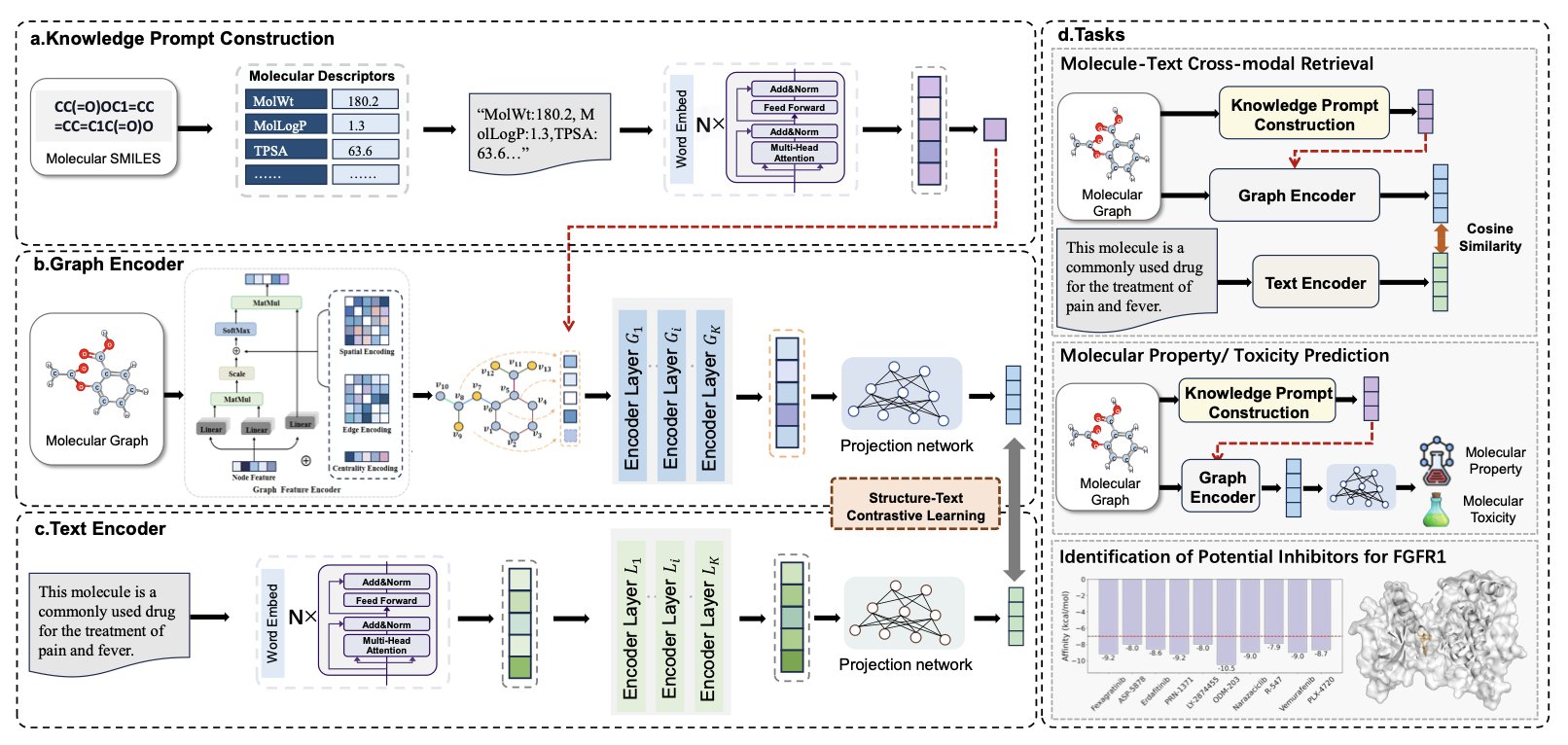
Teaching AI chemistry is like teaching a child to read picture books. We show the AI a picture of a molecule’s structure and a text description, and it learns to connect the two.
This multi-modal learning approach can teach an AI to link images and text, but it doesn’t learn the essentials. It’s like a child who can match the word “apple” to a picture of an apple but doesn’t know that an apple is sweet, crisp, and edible. It has only learned to match patterns.
A new framework called MolPrompt shows a way to teach AI to truly understand chemistry.
AI Also Needs a “Study Guide”
Its idea is to add a module to existing frameworks that aligns with a chemist’s intuition.
It gives the AI a “study guide.”
Here’s how it works:
During training, MolPrompt gives the AI not just the molecule’s graph and name, but also an extra “knowledge prompt.”
This prompt is derived from classic molecular descriptors, such as the logP (lipid-water partition coefficient), molecular weight, and the number of hydrogen bond donors and acceptors. MolPrompt automatically converts these physicochemical parameters, which describe a molecule’s core properties, into natural language.
For example, it might generate a sentence like this: “This is a molecule with a medium molecular weight, moderate lipophilicity, and three hydrogen bond acceptors.”
From a Two-Point Line to a Trinity
The AI’s learning task changes from a simple “image-text” pairing to an “image-text-knowledge” trinity.
The AI must understand that a molecule’s structural graph and its text description both stem from its intrinsic physicochemical properties.
This is like teaching a child about an apple. Besides looking at pictures and reading the word, you also have them take a bite. The child only truly understands what an apple is when the picture, the word, and the “sweet, crisp taste” are all linked together.
Does This Thing Work?
Once it understood the “taste” of chemistry, the AI’s performance improved.
In tasks like predicting molecular properties, assessing toxicity, and cross-modal retrieval, MolPrompt performed better than advanced models that didn’t have a “knowledge prompt.”
More importantly, in a practical test, researchers used MolPrompt to find new inhibitors for the cancer target FGFR1 and successfully identified potential candidate molecules.
This shows that the chemical knowledge MolPrompt learns can be translated into effective insights in drug discovery, not just better scores on academic datasets.
The future of AI-assisted drug discovery may not rely only on bigger models and more data. How we effectively teach AI the first-principles-based chemical knowledge that human scientists have accumulated is just as critical.
📜Title: MolPrompt: Improving multi-modal molecular pre-training with knowledge prompts 📜Paper: https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf466/8240326 💻Code: https://github.com/catly/MolPrompt