
Table of Contents
- MD-LLM-1 isn’t a “camcorder” for traditional molecular dynamics. It’s more like a “director” that can imagine a protein’s “unseen” conformations, leaping over huge energy barriers to see the landscape on the other side.
- This method cleverly uses machine learning to “turbocharge” expensive quantum chemistry calculations, making accurate binding free energy calculations for metal-based drugs accessible for the first time.
- MolGlueDB systematically organizes and provides open access to the most comprehensive molecular glue degrader data to date, offering a much-needed, one-stop platform for researchers in this emerging field.
1. AI Tackles Molecular Dynamics: Predicting the Other Side of Proteins
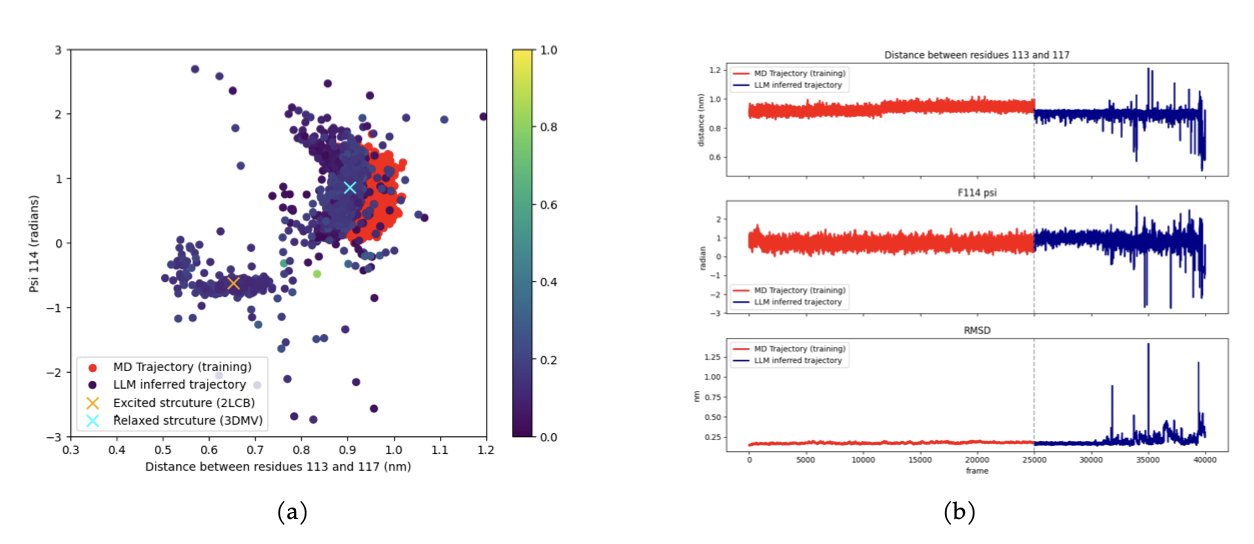
Molecular dynamics (MD) simulations are like trying to find a specific valley in a vast, rugged mountain range while blindfolded. You can only take one step at a time, and you can easily get stuck in a small ditch (a local energy minimum). You could spend millions of CPU hours and still never make it over the highest peak to see the broader landscape on the other side.
MD-LLM-1, the new method in this paper, is like giving you a drone that can teleport.
The researchers used a technique called FoldToken to translate a protein’s 3D structure into strings of “words” that an AI can understand. Then, they used a Large Language Model (Mistral 7B, fine-tuned with LoRA) to act as an “author,” writing the next most logical “sentence” (the next conformation) based on the context.
This AI author isn’t just mechanically repeating stories it has read. Its ability to generalize is what’s truly impressive. The researchers ran a tough test: they trained the model using only data from the “ground state” of T4 lysozyme—its most common, stable form. Then they let the model “create” freely. The model generated not only the ground state but also the “excited state”—a higher-energy, harder-to-reach conformation that is functionally critical. The reverse was also true: training on the excited state allowed it to find the ground state.
This shows that the model learned the “physical grammar” of protein conformational changes, not just a specific trajectory. It understood the underlying logic of transitioning from one state to another, so it could bypass the enormous energy barrier between them and tell you directly what the other side of the mountain looks like.
Many diseases and drug-binding events are linked to these rare, transient “excited states” of proteins. Capturing them with traditional MD is as hard as trying to photograph a bullet as it leaves the barrel. MD-LLM-1 offers a new and potentially much cheaper approach.
Of course, the model is still a specialist, not a generalist. It’s trained for a specific system and isn’t yet a universal tool you can download and apply to any protein. But the possibility it demonstrates is a breakthrough. It points to a future where we can have an AI directly “imagine” and “generate” the most important functional conformations, instead of just waiting for a simulation to get lucky.
📜Title: MD-LLM-1: A Large Language Model for Molecular Dynamics 📜Paper: https://arxiv.org/abs/2508.03709
2. AI Turbocharges Quantum Calculations for Accurate Binding Free Energy
Calculating binding free energy is the ultimate goal—and the ultimate pain—for anyone in computational drug design. It’s the “holy grail” of computational chemistry. Whoever can calculate it quickly and accurately holds the key to predicting a drug’s activity.
We usually have two tools. One is the classical molecular mechanics (MM) force field. It’s fast, but it’s also crude, especially when it encounters atoms it doesn’t “know,” like transition metals. When it sees them, it’s completely lost because its parameter library is empty. The other tool is quantum mechanics (QM). It’s precise, but it’s so slow that you could wait forever. Using it to calculate the free energy of a whole protein is practically impossible.
So, we have QM/MM, a compromise: use QM for the critical region (like the ligand and binding pocket) and MM for everything else. But that’s still not enough. For calculations like free energy perturbation (FEP), which require massive sampling, even QM/MM is too slow.
This study developed a machine learning (ML) potential, an AI model trained specifically to learn the behavior of that small but critical QM region.
It doesn’t need to be fed millions of pre-calculated QM/MM data points. It uses “active learning.” This is like hiring a brilliant intern. You don’t have to teach them everything step-by-step. You let them run the simulation, and when they encounter a conformation they’ve never seen and feel “uncertain” about, they stop and ask, “Boss, I haven’t seen this situation before. Can you run the QM/MM calculation for me?” You only need to answer that one question. They learn from it immediately and then continue their work with confidence.
Through this “on-demand” questioning, a task that would have required thousands of expensive QM/MM calculations was reduced to just a few hundred.
The true power of this method is shown with the GRP78–NKP1339 example. NKP1339 is a ruthenium (Ru)-containing anticancer compound. Anyone who has done molecular simulations knows that handling ruthenium in a classical force field is a nightmare. But this ML/MM method effortlessly produced a binding free energy that agreed well with experimental results.
A whole class of drug targets we couldn’t handle before because they contained metals—like various metalloproteinases—now has a reliable and feasible computational evaluation tool. We no longer need to rely on guesswork or fudge factors.
📜Title: Machine Learning-Enhanced Calculation of Quantum-Classical Binding Free Energies 📜Paper: https://doi.org/10.1021/acs.jctc.5c00388
3. MolGlueDB: The First Dedicated Database for Molecular Glue Degraders
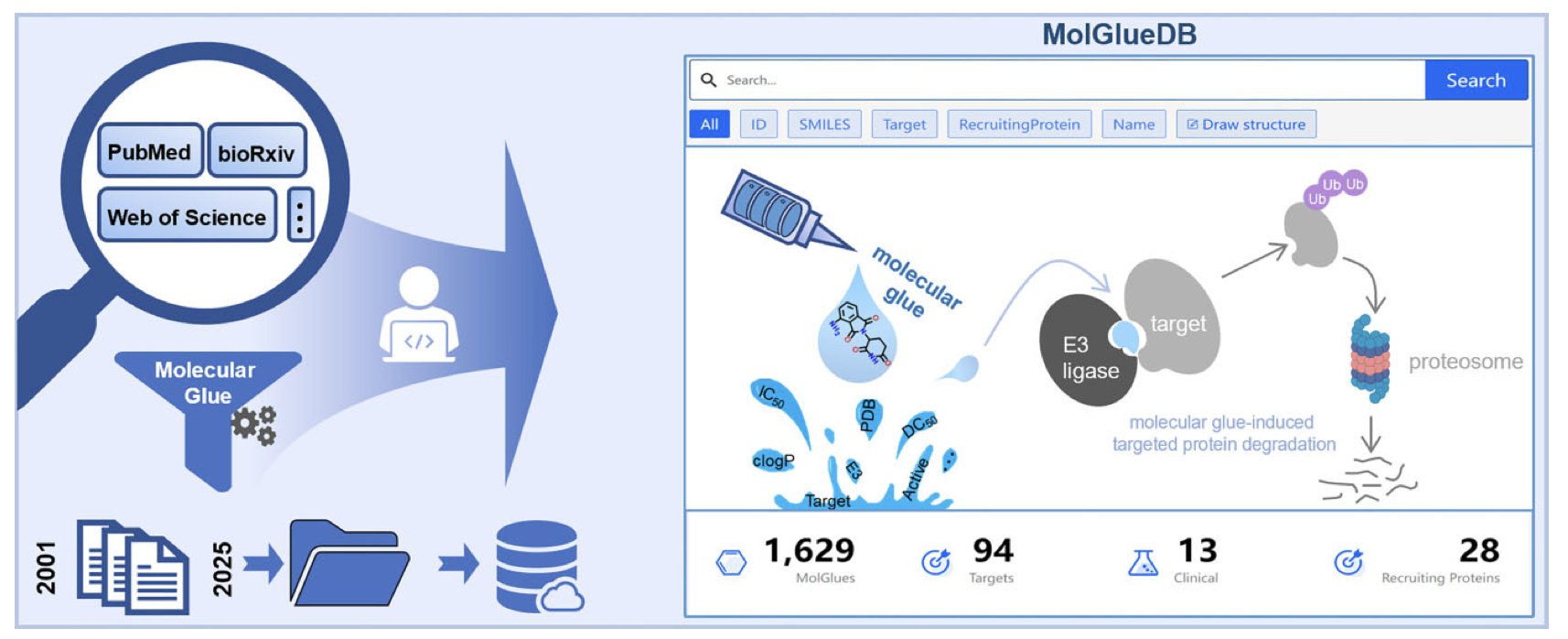
In the field of targeted protein degradation (TPD), PROTAC technology is already well-known, but another branch—molecular glues—is gaining more and more attention.
Like PROTACs, molecular glues use our cells’ built-in “trash disposal system” (the ubiquitin-proteasome system) to degrade disease-causing proteins. But they work more subtly. They don’t need a long linker to “tie” the target protein to an E3 ligase. A molecular glue is more like a small “magnet.” It induces a conformational change on the surfaces of the target protein and the E3 ligase, making two proteins that previously ignored each other suddenly “attracted.” This attraction leads to the formation of a ternary complex, which kicks off the degradation process.
This mechanism gives molecular glues great potential for drugging targets that are difficult for traditional small molecules and PROTACs, like transcription factors without obvious binding pockets. The “rebirth” of thalidomide is a classic example of a molecular glue at work.
A Field Facing Data Silos
As the molecular glue field has grown, related research papers and patents have popped up everywhere. This has created a new problem for researchers: information overload and fragmentation.
Company A reports a new scaffold in a patent, University B publishes a new mechanism in a paper, and Lab C mentions a failed case in a conference abstract. This valuable data is scattered across the internet, creating “data silos.” If we want to systematically understand which E3 ligases have been used, which successful molecular glue scaffolds exist for a specific target, or what the structure-activity relationship (SAR) looks like, we have to spend a huge amount of time and effort.
The MolGlueDB online database was created to solve this problem.
What Does MolGlueDB Offer?
It’s like a central library built specifically for the molecular glue field. 1. Comprehensive Collection: It has compiled over 1,800 entries from 241 publications, covering more than 1,600 different molecular glues. These molecules involve 28 different E3 ligases (recruiter proteins) and 94 different degradation targets. 2. Rich Information: For each molecule, the database provides detailed information, including: * Chemical structure: This is what medicinal chemists care about most. * Biological activity data: Binding affinity (Kd, IC50) and degradation ability (DC50, Dmax). * Physicochemical properties: Molecular weight, cLogP, etc. 3. Powerful Search Functions: In addition to standard text search, it supports “chemical structure search.” This means you can draw a chemical scaffold you’re interested in and have the database find all molecular glues containing that scaffold. This feature is immensely valuable for designing new molecules and analyzing patents. 4. Inclusion of “Negative” Cases: This is a great feature of the database. It includes not only molecules with degradation activity but also “negative” data from molecules that were tested but showed no activity. These “failed” cases are just as important for understanding why a molecule doesn’t work as why it does.
The database also includes some PROTAC molecules that exhibit molecular glue-like behavior, highlighting the connection between the two technologies.
MolGlueDB transforms scattered information into a systematic, searchable knowledge base. It will undoubtedly accelerate innovation across the entire field.
📜Paper: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkaf811/8239508