
Table of Contents
- FRATTVAE rejects atom- or graph-based views, forcing AI to think like a chemist by using “fragments” and organizing them into tree structures. This makes it smart and efficient when handling large, complex molecules.
- AgentD attempts to bundle the tedious computational tasks of early drug discovery into an LLM-driven automated workflow. The idea is neat, but the true test lies in the standalone performance of each module and their combined chemical intuition.
- A new method called VN-EGNNrank uses ligand information to reverse-engineer the location of binding sites on a protein.
1. AI Assembles Molecules Like a Chemist: Building with LEGOs
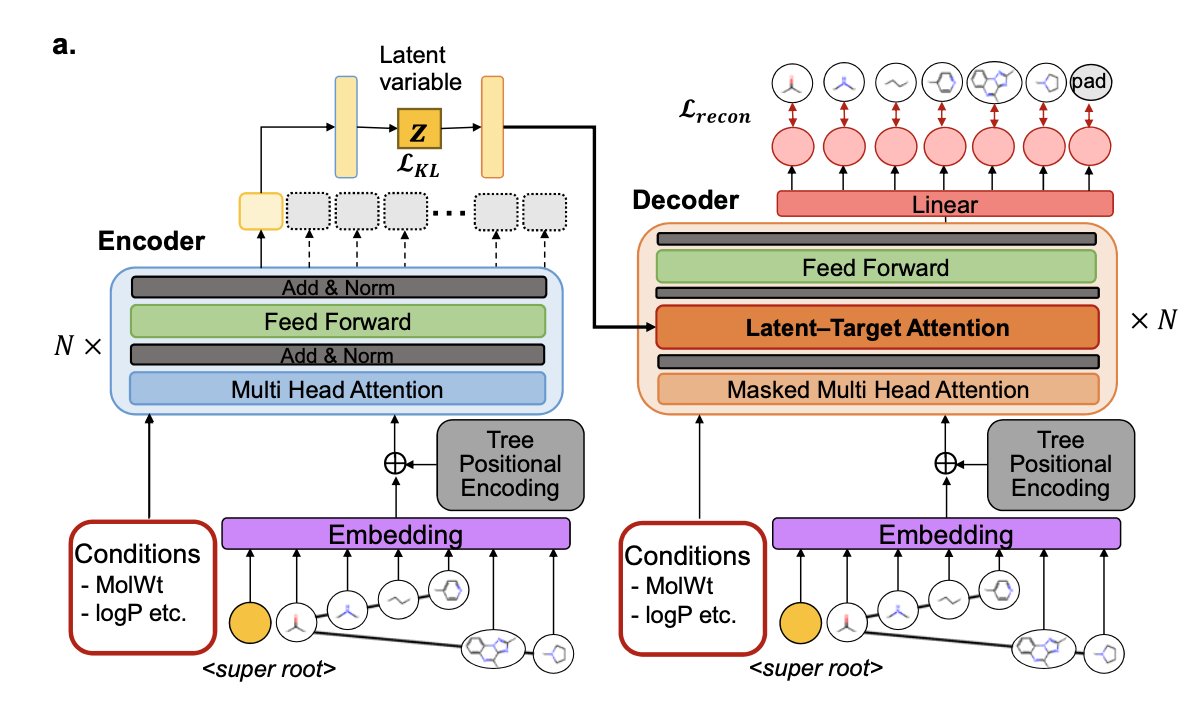
Most molecule generation models swing between two extremes. On one end, you have models based on SMILES strings. They treat molecules like a line of text. They’re simple and fast, but they often produce chemically nonsensical “gibberish.” On the other end are models based on graph neural networks. They see molecules as a map of atoms and bonds. They’re more rigorous, but when dealing with large molecules or complex ring systems, the computation can take forever.
FRATTVAE tries to find a third way. Its approach is a lot like how an organic chemist thinks.
When we see a complex natural product, we don’t count the carbon and hydrogen atoms one by one. We immediately see an indole ring, a sugar group, a lactone ring… We see meaningful “building blocks” like “functional groups” and “scaffold fragments.” FRATTVAE does exactly this. First, it breaks down molecules into these fragments that chemists are familiar with. Then, in its most clever step, it organizes the relationships between these fragments into a “tree structure.”
A tree structure naturally contains hierarchical relationships. The AI doesn’t just know which building blocks are in the molecule; it also knows which one is the main trunk, which are the branches, and how they “grew” step-by-step. To help the AI understand this “molecule tree,” the researchers used a Transformer architecture, which excels at processing sequences and their complex dependencies.
So, how does this AI, armed with a chemist’s mindset, perform?
First, scale. The researchers trained it on a massive dataset of 12 million molecules, resulting in a huge model with one billion parameters. This is quite rare in academia. When you feed a model with data at this scale, it learns more than just a few specific molecular families; it develops a deep, statistical “intuition” for the vast chemical space.
Second, performance. In various benchmark tests, whether it was mimicking the distribution of known molecules (distribution learning) or optimizing molecular properties under given conditions, FRATTVAE blew past previous state-of-the-art models. Its advantage was especially clear when handling large molecules and polymers.
For drug discovery, this means we finally have a tool to more reliably explore the complex chemical space beyond traditional small molecules, like natural products, macrocycles, and even peptides. And it’s faster. This makes high-throughput generation and evaluation of these complex molecules truly feasible for the first time. FRATTVAE isn’t just another VAE model; it might be a new, more powerful way of thinking about chemistry.
💻Code: https://github.com/slab-it/FRATTVAE
📜Paper: https://www.nature.com/articles/s42004-025-01640-w
2. AgentD: An AI Drug Discovery Pipeline. Can It Really Do It All with One Click?
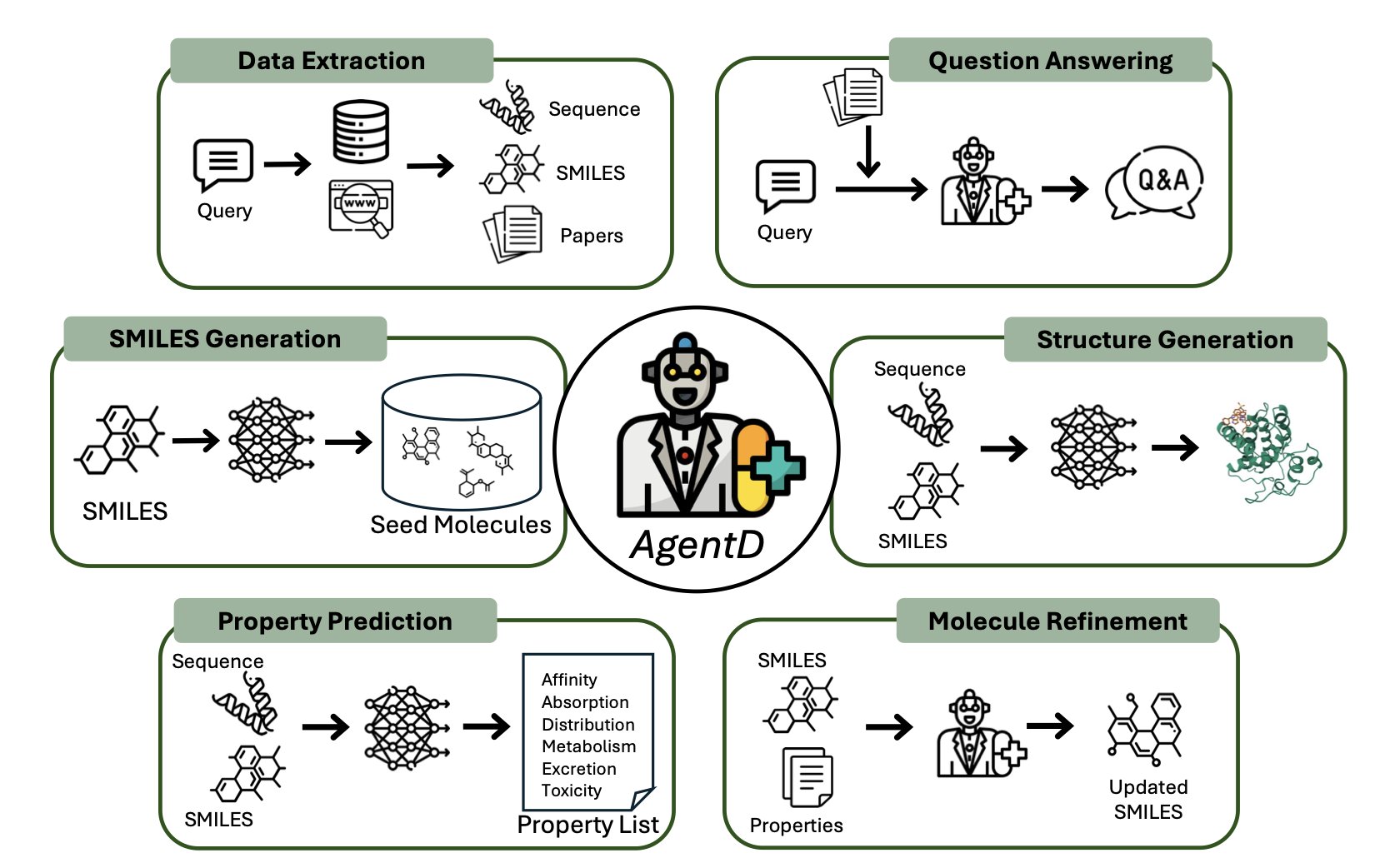
Do you feel like a data courier, switching between different databases and software all day? One minute you’re looking up a FASTA sequence, the next you’re finding a SMILES string, then you open another program to run a prediction… Every computational chemist knows this routine.
What if a Large Language Model (LLM) could handle all of it for you?
This framework, AgentD, aims to be the “general commander” for the early stages of drug discovery. It covers everything from literature search and target information queries to de novo design of new molecules, property prediction, iterative optimization, and even generating the 3D binding mode.
AgentD’s core is an LLM brain, but it doesn’t do the work directly. Instead, it directs a team of “expert” tools. It uses Retrieval-Augmented Generation (RAG) technology, so when asked a technical question, it first “looks up the facts” in specified biomedical databases before answering. This makes its responses more reliable than general-purpose LLMs that are known to hallucinate.
Its molecule generation and optimization capabilities are particularly notable. The researchers tasked it with working on the BCL-2 target in lymphoma. It first used models like REINVENT and Mol2Mol to generate a batch of seed molecules. Then it started predicting up to 67 ADMET properties and binding affinities. Then came the most critical step: iterative optimization.
In their case study, after two rounds of optimization on a virtual library of 194 molecules, the number of molecules with a QED (Quantitative Estimate of Drug-likeness) score above 0.6 increased from 34 to 55. The number of molecules satisfying at least four of the “Rule of Five” criteria increased from 29 to 52. This improvement looks decent. But it acts more like an efficient “filter” and “fine-tuner” than an “alchemist” that can create groundbreaking molecules from scratch. It can polish a pile of rocks to make them smoother, but whether it can turn those rocks directly into gold is still a question.
Finally, it integrates tools like Boltz-2 to generate 3D protein-ligand complex structures, giving you a quick assessment of binding affinity. This is useful for rapidly screening and prioritizing hundreds or thousands of molecules, but don’t expect it to replace precise calculations or crystal structures.
AgentD’s biggest selling point is its “modularity.” The quality of the framework depends entirely on the level of the “expert” tools it integrates. If the tools themselves are not good, AgentD is just for show. But this “plug-and-play” design is smart, allowing for easy replacement with better tools to keep up with the times.
AgentD is an interesting experiment that organizes the AI drug discovery “toolbox” in a very neat way.
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.02.662875v1
3. A New Way for AI to Find Targets: Guiding Molecules to the Right Pocket
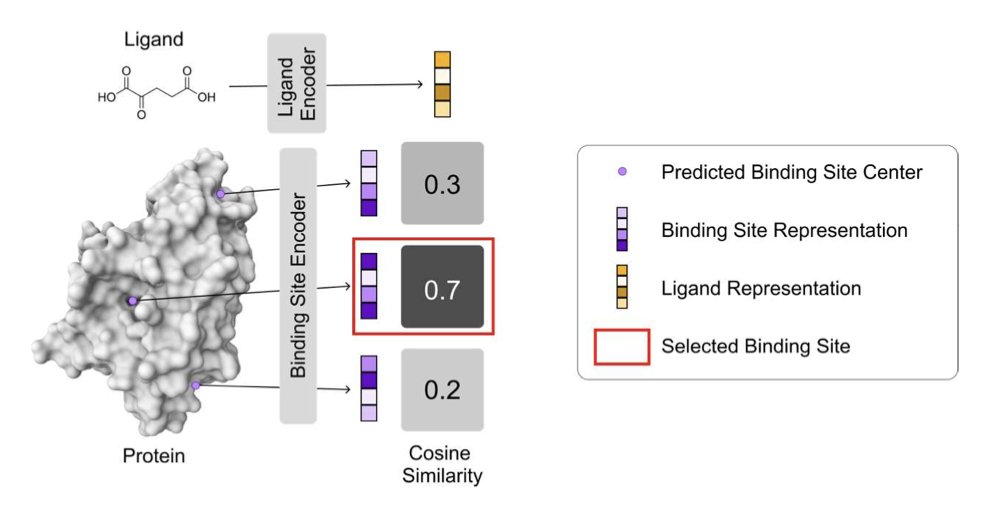
Traditional pocket prediction algorithms are like blind flies, bumping around on the surface of a protein. They tell you “there’s a pit here, a crevice there,” but they don’t care what your molecule looks like or if it can even fit. How is that any more efficient than finding a needle in a haystack?
VN-EGNNrank aims to give that blind fly a GPS. Its core idea is simple but effective: you want to find a pocket for this specific molecule, right? So why not just tell the model about the molecule?
The researchers used a powerful geometric deep learning architecture (VN-EGNN) to “read” the 3D structure of the protein, acting like an experienced building surveyor. At the same time, they used a simple network to create a profile of the ligand molecule.
Then, they used a training strategy called Contrastive Learning. It works hard to pull the correct “molecule-pocket” pairs closer together while pushing the mismatched ones far apart. After this training, the model learns what kind of molecule likes what kind of pocket, gaining the “chemical intuition” that we chemists dream of.
The results on several standard datasets show that this little model, with only 3 million parameters, can go toe-to-toe with the 25-million-parameter heavyweight DiffDock, sometimes even outperforming it. It’s like a go-kart posting a Formula 1 lap time. For people running large-scale virtual screens every day, this means we can screen libraries in days or even hours that used to take weeks, with reliable results.
We know that most current AI models for pocket prediction have a “path dependency”—they are especially good at finding the “classic” orthosteric sites, which are located at the active center and are deep and large. But the future of drug discovery largely depends on the more hidden and tricky allosteric sites, because they often offer finer control and fewer side effects.
The researchers found that when a protein has multiple ligands (often including allosteric modulators), the performance of traditional models plummets. But VN-EGNNrank has a natural advantage here because it considers the ligand itself. It’s not guessing blindly; it’s finding the right “lock” for a specific “key,” and that “key” could open the front door or a side window.
Of course, the authors admit that the model still doesn’t handle cases where one ligand corresponds to multiple pockets very well, and the training data still has too few allosteric sites.
📜Title: LIGAND-CONDITIONED BINDING SITE PREDICTION USING CONTRASTIVE GEOMETRIC LEARNING
📜Paper: https://openreview.net/forum?id=86TgAWEkaD