
Table of Contents
- One paper makes a compelling case that in genomics, simply extracting a model’s “embeddings” is just as effective as—and much faster and cheaper than—brute-force fine-tuning.
- The new FGBench benchmark shows that today’s most powerful AI models perform like failing undergrads when it comes to understanding the basic language of chemists: functional groups.
- What makes the “Baishenglai” (BSL) AI platform stand out isn’t its list of features, but the fact that it actually found new, active molecules in a lab, distinguishing it from 99% of AI drug discovery stories.
1. Genomics AI: Embeddings Are King, So Why Fine-Tune?
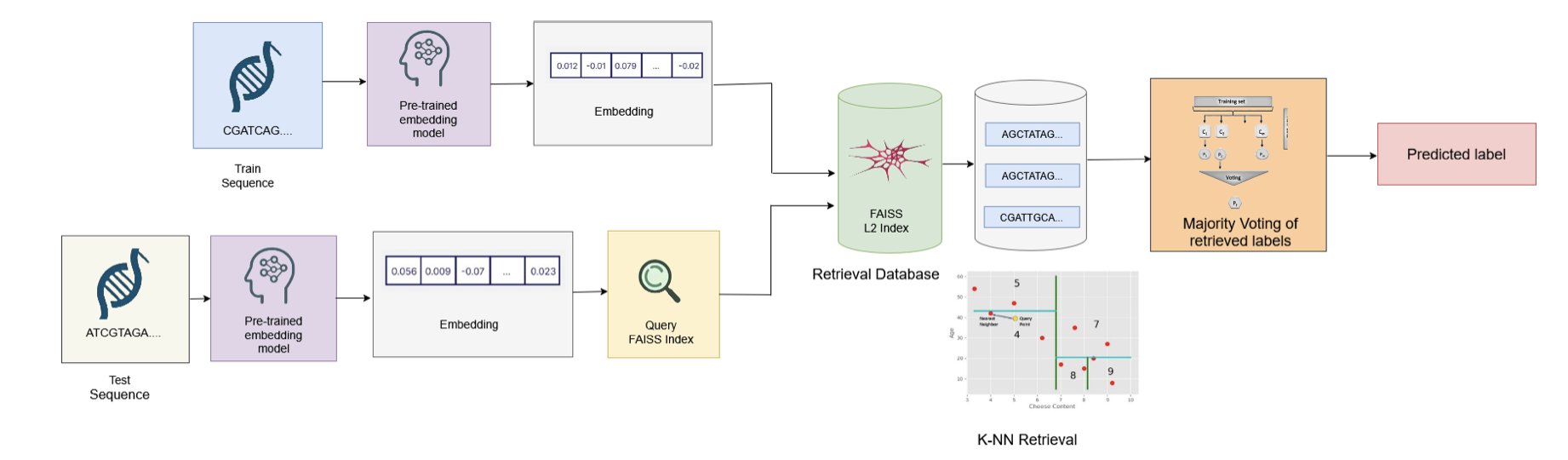
The paper’s title, “Embedding is (Almost) All You Need,” is full of provocation. It’s a clear nod (or maybe a jab) at “Attention is All You Need,” the paper that kicked off the Transformer era. This new work is a declaration of thrift for the field of genomics AI.
We’ve all fallen into a pattern: get a genomics problem, find the biggest pre-trained DNA model available, and then “fine-tune” it with our own data.
This process is like hiring a Nobel laureate in physics to teach your kid elementary school math, and then putting the physicist through months of “elementary teaching methods” retraining. It’s absurd, expensive, and painfully slow. Every time a new problem comes up, you have to put the poor professor through the wringer all over again.
This paper’s method is more like a pragmatic project manager. It says we don’t need to retrain the Nobelist at all. We just need to ask him, “Professor, based on your life’s work, please summarize the core points of this physics problem in a few sentences.” Those few sentences are the “embedding.” Then, we hand these valuable “core points” to a college student (a lightweight classifier) and let him grade the elementary math tests.
And it turns out the student does a great job, fast.
The data shows that this “embedding + lightweight classifier” combination not only performs on par with the massive effort of fine-tuning but completely dominates it in efficiency. Inference time is cut by 88%, and the carbon footprint drops by over 8 times. For people fighting with compute resources and project deadlines every day, this means we can run experiments in a single day that used to take a week, and for less money.
Even more important is its “generality.” A single set of embeddings extracted from the HyenaDNA model could handle nine completely different tasks, like enhancer classification and species identification, and performed reasonably well on all of them. This shows that these pre-trained models have truly learned some universal “knowledge” about the language of DNA. We just didn’t realize that using this “knowledge” directly was powerful enough on its own.
Fine-tuning still has its place for tasks that demand extreme performance and have huge amounts of data. But this paper sets an affordable baseline. From now on, anyone claiming their model needs fine-tuning must first answer the question: “How much better is your result than just using embeddings? And is that tiny performance gain worth ten times the cost?”
📜Title: Embedding is (Almost) All You-Need: Retrieval-Augmented Inference for Generalizable Genomic Prediction Tasks 📜Paper: https://arxiv.org/abs/2508.04757v1
2. AI Understands Chemistry? The FGBench Exam Makes Large Models Flunk
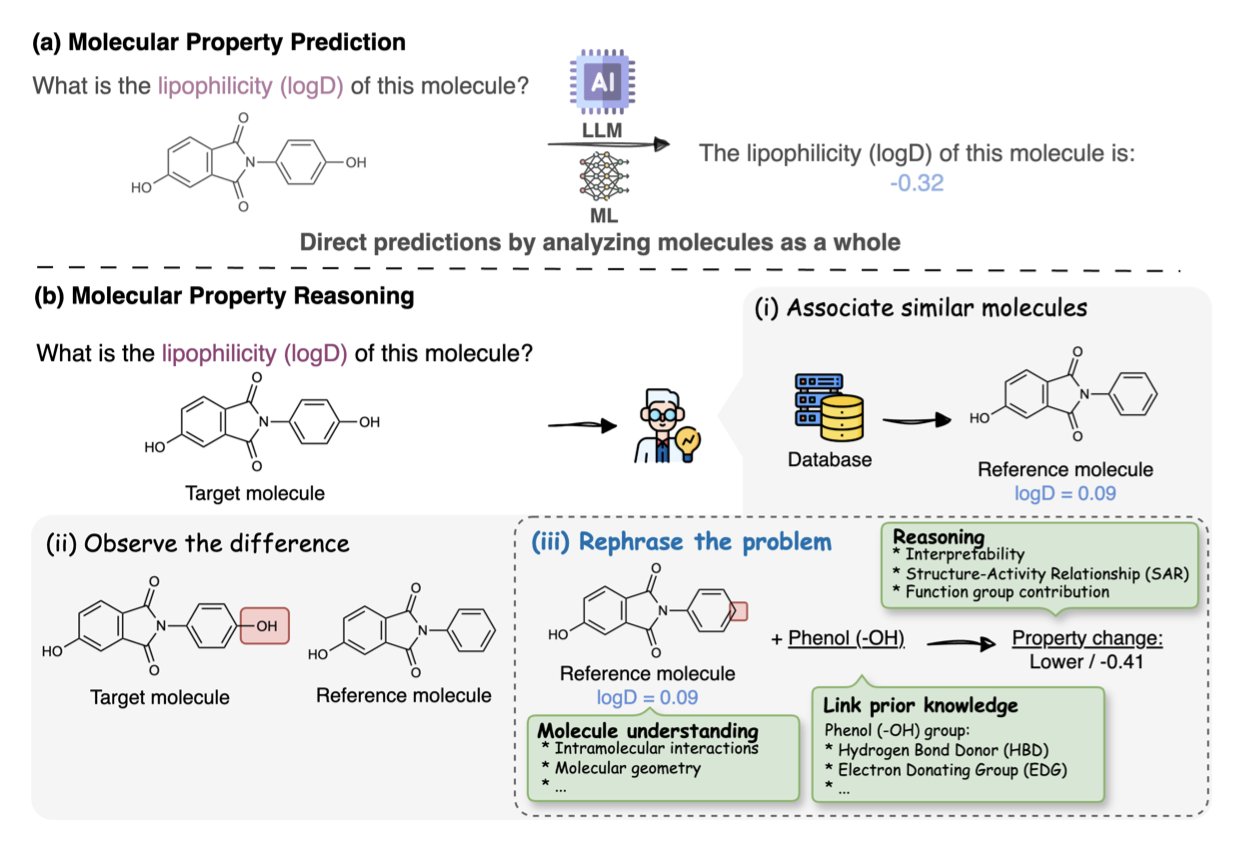
For chemists, functional groups are our alphabet. When we see a molecule, we don’t just see a diagram of connected atoms and bonds. We immediately see hydroxyls, carboxylic acids, amides, Michael acceptors. The combination of these functional groups defines a molecule’s personality, its reactivity, and whether it could become a drug.
But today’s Large Language Models (LLMs), even one as powerful as GPT-4, are largely “illiterate” when reading molecules. They can process SMILES strings or molecular graphs and memorize vast numbers of molecules and their properties. But if you ask a real chemist’s question—“If I hydrolyze this ester to a carboxylic acid, how will the molecule’s water solubility change?”—it will likely start spouting confident nonsense.
The new FGBench benchmark was created to expose these “pseudo-chemists.”
It doesn’t just collect more molecules and properties. It created a massive question bank of 625,000 “chemical reasoning problems.” Every question is about functional groups. For example, “The difference between molecule A and molecule B is that A’s nitro group is replaced by an amino group. Which is more lipophilic?” This is a true test of chemical understanding.
To ensure the quality of these “exam questions,” the researchers used a “disassemble and reassemble” validation strategy. They made sure that after each functional group modification, the result was still a chemically plausible and structurally complete molecule. It’s like an exam creator doing all the problems themselves first to make sure there are no errors or ambiguities.
So, how did the AIs do on the test?
The results were abysmal. The benchmark showed that even the top LLMs struggled with these functional-group-level reasoning tasks. What does this tell us? It says their so-called “understanding” is more about pattern matching based on superficial correlations than causal reasoning based on underlying chemical principles. They’ve learned to memorize the dictionary but haven’t yet learned to use the words to form sentences and write essays.
What does this mean for drug discovery? We urgently need an AI partner that can converse with us, one that can say, “I think we should introduce a hydrogen bond donor here to improve activity,” not a black box that just spits out numbers. FGBench is the driving force compelling the next stage of AI development to move toward “explainability” and “structural awareness.”
It’s not just a dataset; it’s a new, more rigorous curriculum. With it, we can finally start training and selecting AI models that can actually think in our language.
📜Title: FGBench: A Dataset and Benchmark for Molecular Property Reasoning at Functional Group-Level in Large Language Models 📜Paper: https://arxiv.org/abs/2508.01055v2
3. An All-in-One AI Drug Discovery Machine That Actually Found Active Molecules
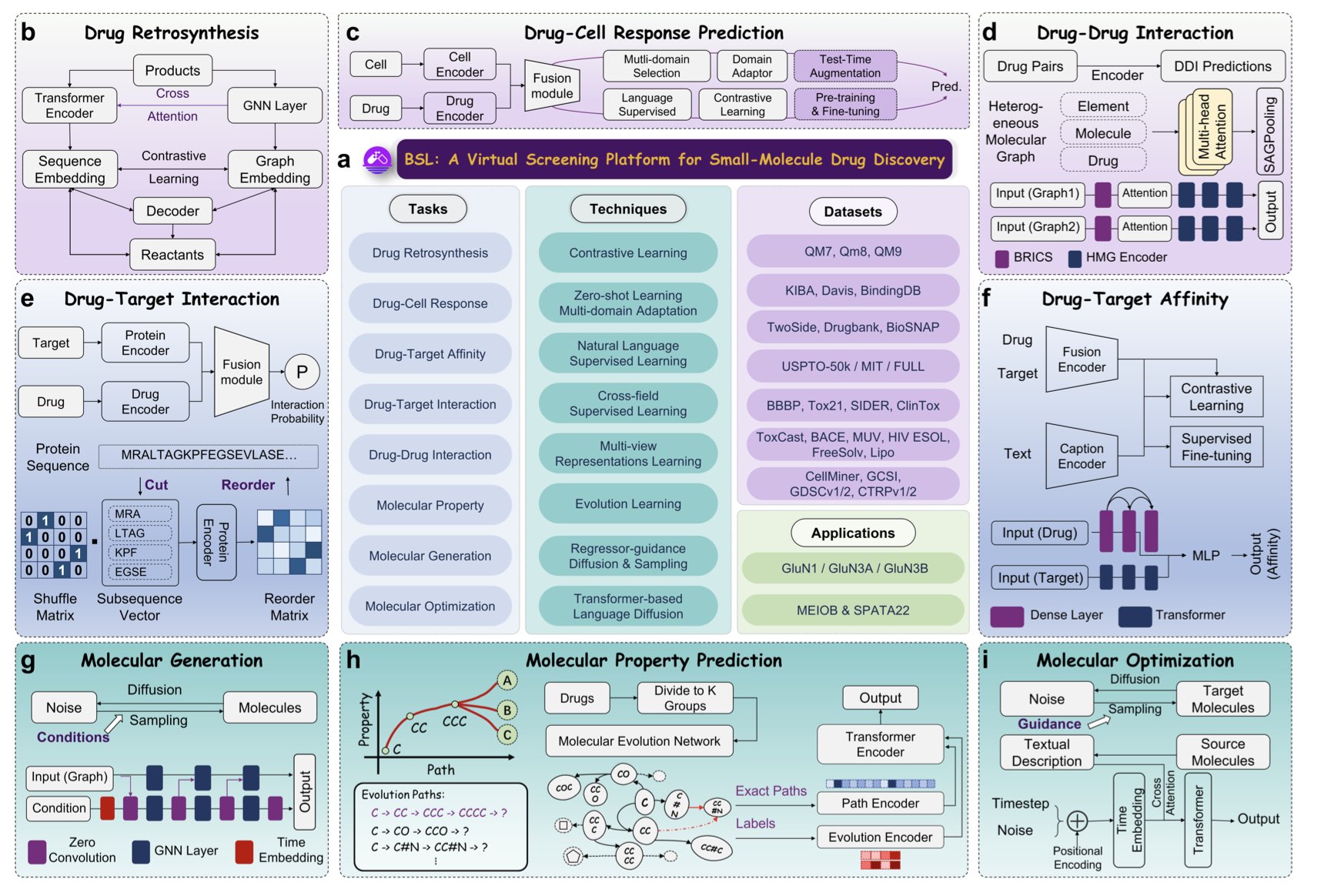
Baishenglai (BSL) presents a grand vision, integrating seven major functions: de novo molecule generation, structure optimization, ADMET prediction, drug-target interaction… basically, any computational step you can think of on a drug discovery flowchart, it aims to cover.
But the most critical part of the paper is a real case study backed by experimental results.
Most papers on AI platforms end with pretty ROC curve charts, telling you how high their model’s AUC score is on some public dataset. That’s fine, but it’s like a race car driver who’s only ever driven in a simulator. You never know what will happen when you throw them onto a real, rain-slicked track at Spa.
The BSL researchers threw themselves onto a real track. They picked a tough target—the GluN1/GluN3A NMDA receptor. This is a difficult target with limited structural information. Then, they used BSL to design new modulators for it.
Here comes the most important step: they actually synthesized the molecules designed by the AI and then took them to a wet lab to test their biological activity in vitro.
And they found three compounds with clear activity.
This step is the dividing line between heroes and talkers. It means BSL isn’t just a better database search engine; it actually possesses a kind of “chemical intuition.” It can explore unknown chemical space and propose sound, experimentally verifiable hypotheses.
The source of this ability likely comes from its focus on the OOD (out-of-distribution) generalization problem. The essence of drug discovery is to find molecules we’ve never seen before that happen to solve our problem. An AI that can only perform well on problems within its “training set” is of limited value to us. BSL was designed from the start to solve this “out-of-the-box” problem. That’s the right path, even if it’s the harder one to walk.
📜Title: Empowering Drug Discovery through Intelligent, Multitask Learning and OOD Generalization 📜Paper: https://arxiv.org/abs/2508.01195