
Table of Contents
- This open-source tool cleverly makes AlphaFold2 “err” on purpose to generate multiple starting points, then uses physical simulations to explore them. This greatly speeds up how we capture snapshots of proteins in motion.
- By breaking down the complex problem of solubility into more fundamental physical processes and tackling each with machine learning, this new method gives us the clearest roadmap yet for predicting solubility.
- This new approach breaks quantum chemistry into pieces and uses machine learning to correct key physical forces. It achieves extremely accurate binding affinity ranking in under 5 minutes—a big step for computational drug screening.
1. AI Gives Molecular Simulations a “Teleporter,” Ending the Slow Crawl
In structural biology, AlphaFold gives us static “photographs” of proteins—clear, beautiful, like a well-made product catalog. But that’s not enough. Proteins in a cell aren’t motionless sculptures. They twist, breathe, and change shape. A drug molecule often binds to a fleeting “active” or “inactive” conformation that you’d never see in a static picture.
To see proteins in motion, we turn to molecular dynamics (MD) simulations.
MD is like a supercomputer that can film a “movie” of the molecular world. The problem is, the playback speed of this movie is slow enough to make you question reality. Running a simulation for a few hundred nanoseconds can tie up a computing cluster for weeks. And you’ll often find that your protein, the star of the show, spends the entire simulation napping in its lowest-energy “comfort zone,” never moving an inch. The “hidden pocket” we actually want to see, the one that binds the drug, might be just over a massive energy “mountain” that’s too high to climb.
So, we’ve been stuck. We have the ability to make the movie, but we can’t afford the time or the cost.
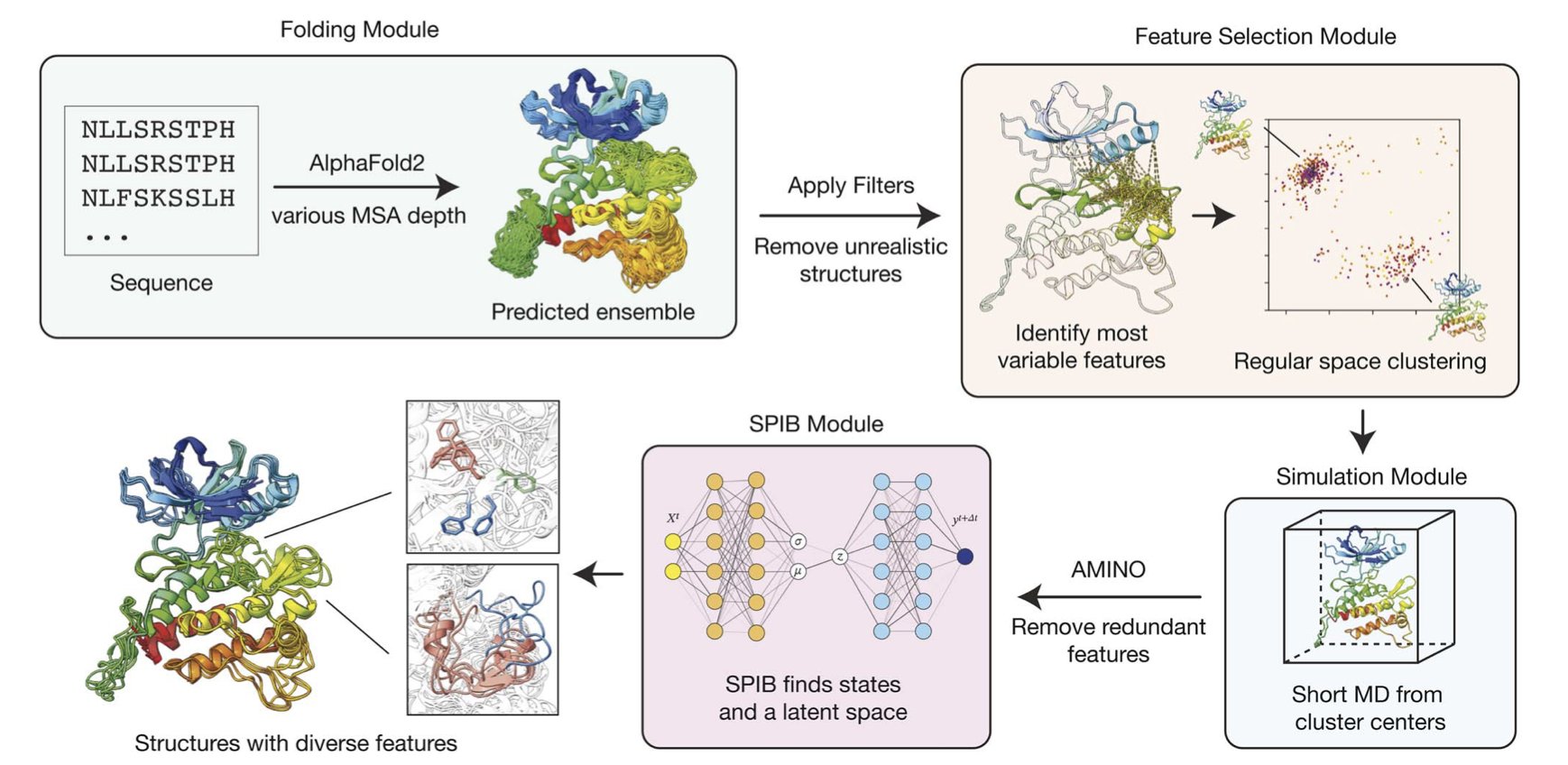
The af2rave tool from this paper gives the film crew a very clever script supervisor and a bunch of teleporters.
Here’s the logic: We know AlphaFold2 is great at predicting structures, but that’s when we feed it a huge amount of Multiple Sequence Alignment (MSA) information. What if we intentionally give it less information? What if we only give it a few clues and let it “guess”? When we do that, AlphaFold2 becomes a bit uncertain. It might propose several different, yet plausible, structures.
This is the core idea of af2rave. It leverages this AI uncertainty. The different starting structures it generates are like explorers placed in different “valleys” on the protein’s energy landscape. Then, af2rave starts a relatively short MD simulation in each of these valleys. It’s like exploring a map from several points at once. Compared to sending a single explorer from one entrance to spend a year climbing mountains, this multi-start strategy maps out the entire landscape much, much faster.
They tested it on a target called DDR1 kinase, a protein known to have different functional states. af2rave found all the known states without much trouble. On the COVID-19 spike protein, its sampling efficiency was on par with very long simulations that require massive computational resources.
The tool is open-source, and some of the complex parameter choices have been automated. Now, a medicinal chemist or biologist doesn’t need a PhD in computational physics to use this powerful tool. They can see if the protein they’re interested in has other “candid shots” besides the “studio portrait” we always see in the PDB database.
Can it find every possible protein conformation?
Of course not. The starting points it finds are still limited by AlphaFold2’s capabilities. If an extremely rare conformation exists that the AI can’t even imagine, then it can’t be simulated. But we’re moving from waiting blindly for long periods to exploring strategically and efficiently.
💻Code:https://github.com/tiwarylab/af2rave 📜Paper: https://pubs.rsc.org/en/content/articlelanding/2025/dd/d5dd00201j
2. AI Predicts Solubility: It’s Physics, Not Magic
Predicting a new molecule’s solubility in water is about as hard as picking next Tuesday’s lottery numbers. We have many computational models, but most are either rough rules of thumb or incomprehensible black boxes. You feed in a molecule, and a number comes out, with accuracy being a matter of luck.
The authors didn’t try to solve the “solubility” equation with one giant, all-encompassing AI model. Instead, their approach is more like that of a seasoned mechanic. He doesn’t just grab a sledgehammer; he first takes the machine apart to see which specific part is broken.
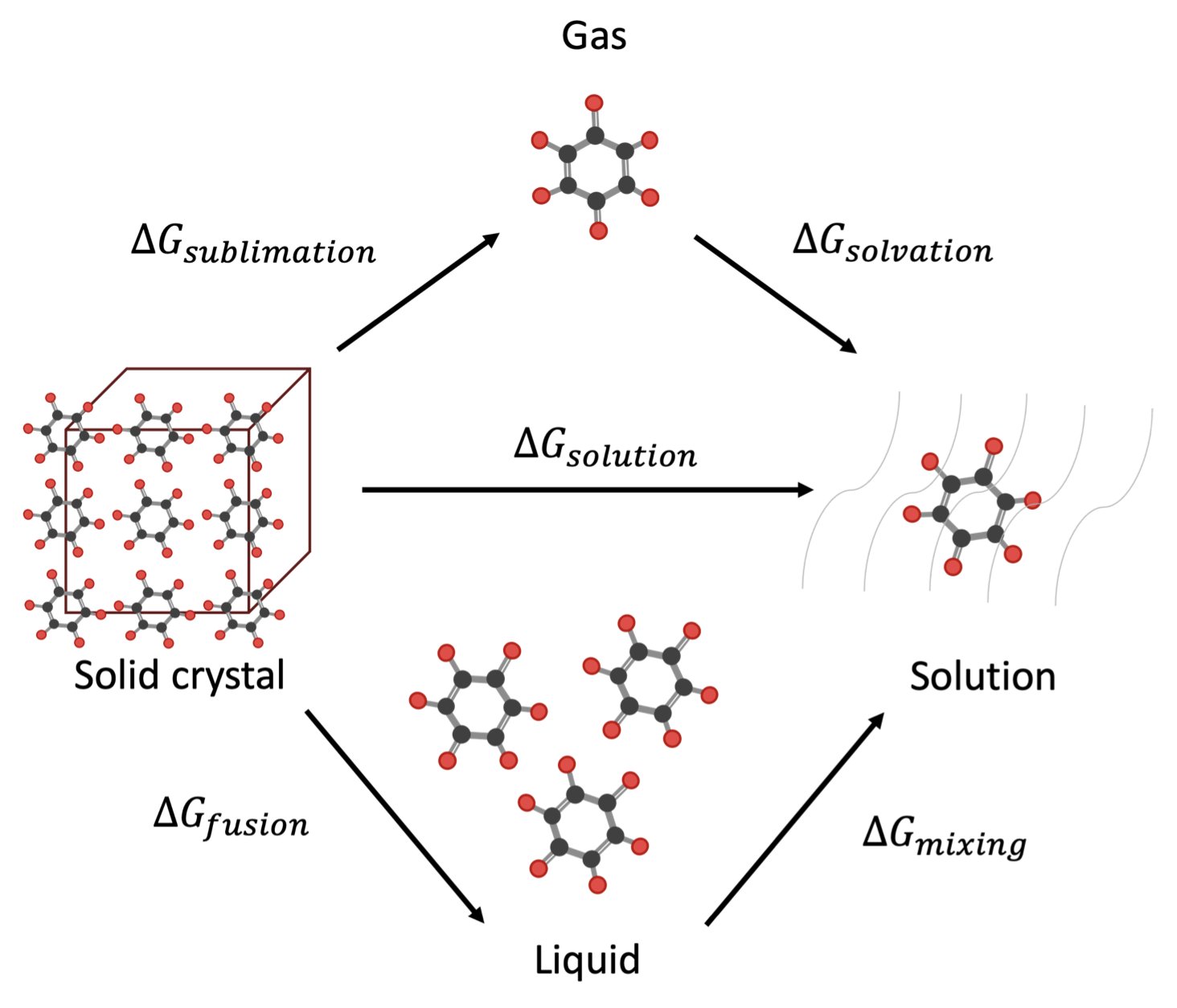
They broke the process of dissolution down to its fundamental thermodynamic struggle. For a molecule to dissolve, it has to make two decisions. First, it must decide to leave its own cozy crystal home. The difficulty of “leaving home” depends on its melting point and enthalpy of fusion. A molecule with a melting point as high as a diamond has an extremely stable crystal structure. The molecules are holding on to each other so tightly that you have no chance of pulling them apart. Second, it has to find the outside world (the solvent) attractive enough. The strength of this attraction is what’s called the “activity coefficient.”
The beauty of this paper is that it uses machine learning models to predict each of these separate physical quantities. Then, it uses a classic thermodynamic cycle equation to combine them and calculate the final solubility. It’s a very smart “divide and conquer” strategy.
And there’s a technique here called “solvent ensembles” that is just brilliant. Here’s the logic: Let’s say I already know my molecule’s solubility in water, and now I want to predict its solubility in ethanol. The traditional way is to start from scratch and calculate everything again. This new method says, “Wait, we already have valuable experimental information about this molecule. Let’s not waste it!” It uses the known data in water as a strong “anchor” to calibrate and predict its behavior in ethanol. This greatly improves the prediction’s robustness and accuracy because it uses all available information.
On a huge dataset with over a hundred thousand experimental data points, the method performed quite well, especially in its ability to predict solubility at different temperatures, something many older methods struggle with. It also happens to predict the oil-water partition coefficient (logP) without having been specifically trained on logP data. This strongly suggests the model might be doing more than just curve-fitting; it may have actually learned some underlying, universal principles of physical chemistry.
The model still depends on high-quality training data, and experimental data for things like melting points and enthalpies can be unreliable. Can it handle the weirdly shaped, highly charged, and very flexible “ugly” molecules we see in our daily work? That remains to be seen.
📜Title: Accurately Predicting Solubility Curves via a Thermodynamic Cycle, Machine Learning, and Solvent Ensembles 📜Paper: https://doi.org/10.26434/chemrxiv-2025-jd8zw
3. Fast and Accurate AI for Drug Discovery: Ranking Binding Affinity in 5 Minutes
How do we pick the one true partner for our target protein from thousands of candidate molecules? To avoid wasted effort, we turn to computational chemistry, hoping a computer can tell us which molecules have more potential. But the tools we have are either lightning-fast but basically guesswork (molecular docking) or stunningly accurate but so slow you might retire before it’s done (free energy perturbation, or FEP). We’ve always wanted a tool that is both fast and accurate.
Lao and Wang’s paper splits this complex problem into two parts.
First, they use a method called Geometry-Based Molecular-Fragmentation with Density Matrix (GMBE-DM). The name sounds intimidating, but the core idea is simple: how do you eat an elephant? One bite at a time. Trying to calculate a protein-ligand complex with thousands of atoms using high-accuracy quantum chemistry would make a supercomputer cry. So, they “chop up” this large system into countless small, computable fragments and then cleverly piece the results back together. It’s a classic divide-and-conquer strategy.
But that’s not all. Just breaking down the problem isn’t enough to guarantee accuracy. There’s a critical but very tricky component of the forces between a drug and a protein called van der Waals forces, or dispersion forces. You can think of it as the weak, fuzzy attraction between two uncharged fluff balls when they get close. It’s everywhere, yet it’s extremely hard to describe precisely. Many computational methods fail right here.
The masterstroke in this paper is their use of a specialized, physics-based machine learning model (D3-ML) to “patch” this dispersion calculation. This AI model has seen tons of data. It knows when and where traditional calculation methods are likely to get it wrong, and it precisely corrects that error. It’s like an experienced mechanic who can hear an engine and know exactly which screw is loose, then goes and tightens it.
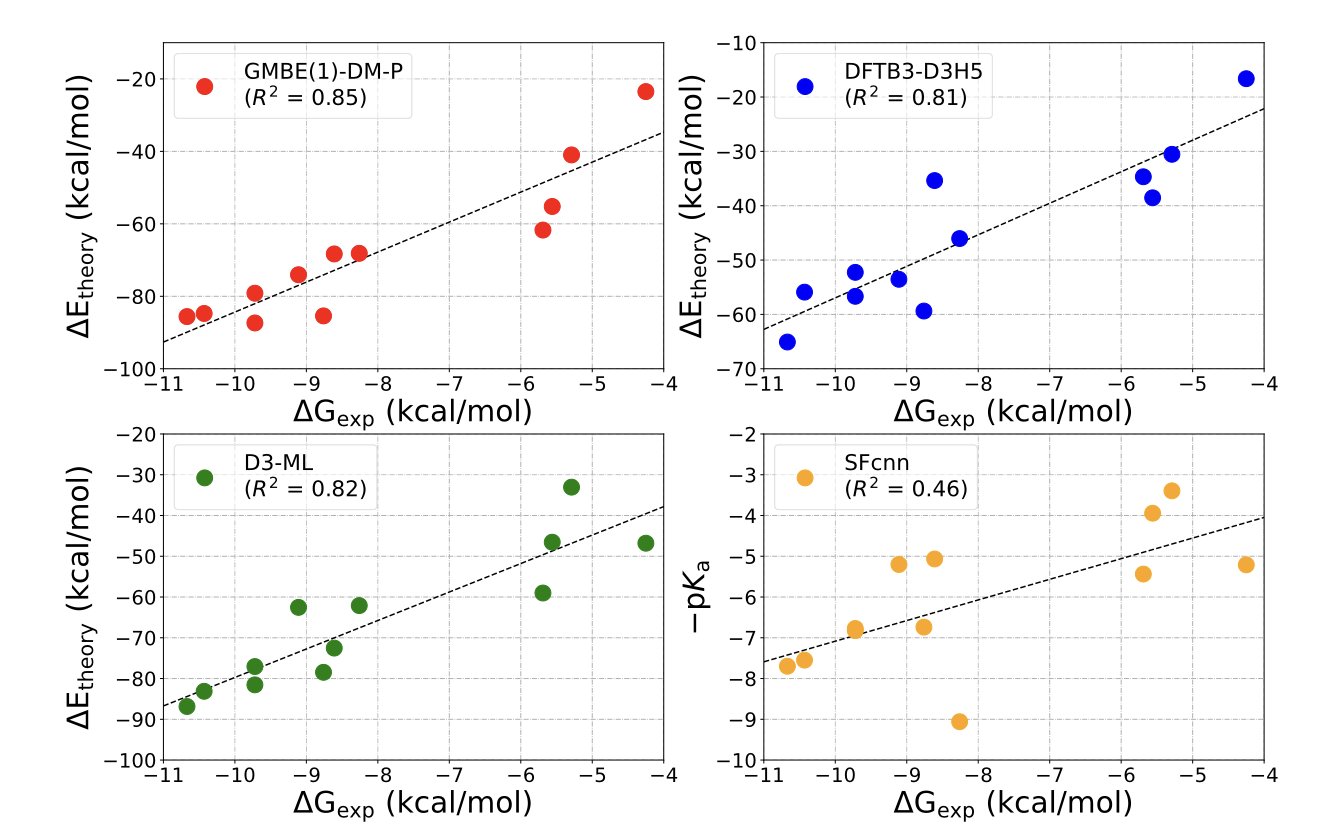
So how did it do? The results are surprisingly good. On several classic kinase targets, this combined approach achieved a correlation (R²) of 0.87 with experimental data when ranking the binding affinities of a series of ligands. In our field, that’s a number that makes everyone sit up and take notice. And a calculation for one complex takes less than 5 minutes. This means we finally have a medium-to-high-accuracy tool that can actually be used at scale in real projects, not just in academic papers.
What’s more interesting is what they found when they deliberately ignored the annoying water molecules from the crystal structures and didn’t even calculate the ligand’s desolvation energy. The ranking accuracy was actually higher. This is a slap in the face to computational chemists. We’ve spent years carefully modeling every single water molecule, trying to figure out what they do. And it turns out the best strategy might be “out of sight, out of mind”? This is another reminder that sometimes, a simpler model that captures the main part of the problem is better than a complex model that tries to do everything but just ends up introducing more noise.
Of course, we need to stay calm. The method was validated on a few fairly “well-behaved” systems. Can it handle targets that are highly flexible and undergo large conformational changes? We don’t know yet. But it points down a very clear path: the future of computer-aided drug discovery lies in the smart combination of first-principles physics models and data-driven machine learning. We might still be far from truly “designing” drugs, but at least now we have a better sieve to pan for gold a lot faster.
📜Title: Accurate and Rapid Ranking of Protein–Ligand Binding Affinities Using Density Matrix Fragmentation and Physics-Informed Machine Learning Dispersion Potentials 📜Paper: https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cphc.202500094