
Table of Contents
- BioTransformer 4.0 uses smart filtering and a modular design to change metabolite prediction from an “ocean of information” into a “practical map” for drug design.
- The BIND framework tries to solve the multiscale bio-network prediction problem, from molecules to phenotypes, with an integrated AI platform. Its real value depends on whether its new leads can be experimentally confirmed.
- An AI framework called CellAtria tries to turn the tedious process of single-cell data analysis into a simple conversation with a chatbot.
1. No More Guesswork in Metabolism Prediction? Meet BioTransformer 4.0
![]()
You have a promising hit compound. The synthesis route is optimized. Then, you put it in an animal, and it either gets metabolized into nothing or turns into something toxic, killing the project. Predicting drug metabolism has always been a thorn in the side of R&D scientists.
Older prediction software was often one of two things. It was either a rule-based machine that spat out a huge list of theoretically possible metabolites, forcing you to search for the important one in a sea of noise. Or it was a black box, giving you no clue how it worked and missing the one critical, “fatal” metabolite that you only discover in late-stage experiments when it’s too late. This is a classic signal-to-noise disaster.
BioTransformer 4.0’s “Metabolite Validation Module” doesn’t just list every chemical possibility. It compares its predicted results against a database of known human metabolites. The key here is its ability to filter out structures that are “theoretically possible but realistically never produced.” According to the paper’s data, this step can reduce the number of predicted results by up to 80%.
What does that mean? It’s like having an experienced DMPK (Drug Metabolism and Pharmacokinetics) expert standing behind you. As you stare at a screen full of predicted structures, they tap you on the shoulder and say, “Don’t bother. This one, this one, and that one look wrong. We’ve never seen that kind of transformation in our lab. Toss them.” Your workload instantly shifts from finding a needle in a haystack to something much more manageable. You can focus your energy on the few high-risk metabolites.
Another interesting feature is the “Abiotic Metabolism Module.”
Anyone in drug development knows that a molecule faces more than just P450 enzymes in the liver. Your molecule might end up in a sewage treatment plant, break down under sunlight, or even react with something in an IV bag. The addition of this new module expands BioTransformer’s scope from just pharmacokinetics to environmental toxicology and even formulation stability. This makes the tool much more versatile for assessing a molecule’s full life-cycle risk.
And then there’s the “Customizable Multi-Step Prediction.”
This is where it gets powerful. Older tools were mostly one-step. You input a parent compound, and they give you a list of possible “child” metabolites. But to find out what the “grandchildren” look like, you had to manually re-enter each child. Now, you can string different metabolic modules together like building blocks. For instance, you can set a molecule to first undergo a phase I oxidation reaction, then feed all the oxidized products directly into a phase II glucuronidation module to see what happens next. This simulation of chain reactions is closer to a drug’s real journey in the body and can help you uncover deeper metabolic pathways.
As for performance, the paper reports recall rates of 87.2% on the DrugBank dataset and 91.6% on the PhytoHub dataset. The numbers look good, meaning it can “see” the vast majority of known metabolites. Of course, no computational tool is perfect; it can’t be 100% accurate. But as a screening tool, it’s good enough.
It allows us to “kill” a bunch of bad ideas before synthesis or flag a structure with potential metabolic risks early on. In the money-pit industry of drug development, discovering a problem even one day sooner saves not just lab supplies, but precious time and opportunity cost.
📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.28.667289v1
2. BIND: Predicting Vast Biological Interactions with a Knowledge Graph
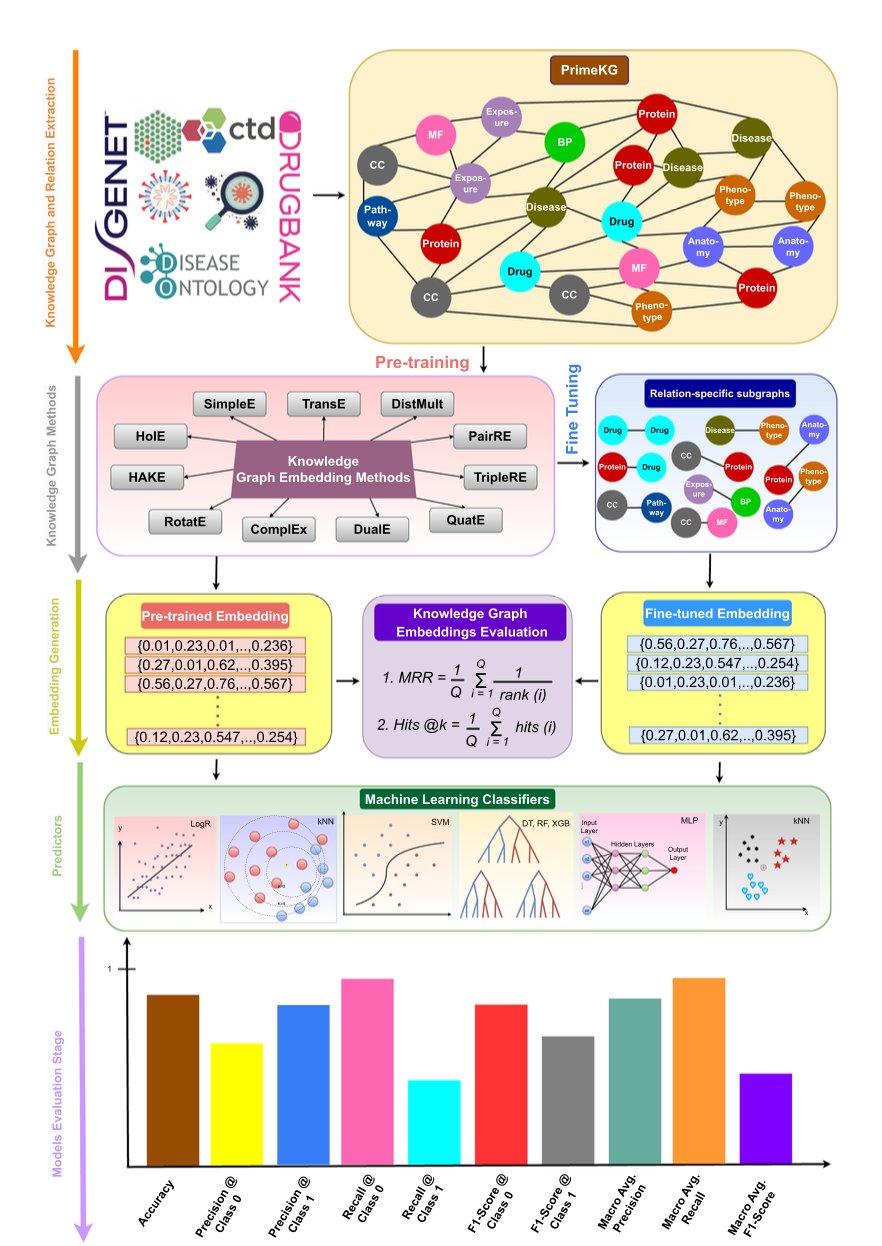
In biology research, especially drug discovery, we are always dealing with a vast, intricate “relationship network.” How do proteins bind? Which genes does a drug affect? What phenotype does a certain gene mutation lead to? Each of these questions is a tough one.
Traditional methods are like a blind person touching an elephant—you can only study one part at a time. Now, the authors of this paper present a tool called BIND, which they claim uses a knowledge graph and machine learning to give us a pair of glasses to see the whole elephant.
The researchers took 11 popular methods and ran them on a massive knowledge graph containing 8 million known interactions involving nearly 130,000 nodes. The knowledge graph is ambitious, incorporating 30 different types of biological relationships, from molecular interactions to links between drugs and clinical phenotypes.
Anyone who has worked on bioinformatics predictions knows that one of the biggest challenges is data imbalance. Positive samples (known interactions) are scarce, while negative samples (unknown or non-existent interactions) are abundant. This can lead to a model that just learns to say “no.”
BIND uses a clever trick: “two-stage training.” In the first stage, they “warm up” the model with a carefully balanced dataset to learn basic interaction patterns. In the second stage, they “toughen up” the pre-trained model by exposing it to real-world, imbalanced data. The method isn’t new, but it is effective for improving performance on classic problems like protein-protein interactions (PPIs).
They report impressive F1 scores, with many tasks reaching 0.85 to 0.99, which sounds almost perfect. But those who have been in the trenches know the devil is in the details. These high scores were achieved on specific test sets. The real test is how many of its predicted “new” interactions are biologically meaningful discoveries versus statistical illusions from model overfitting.
The authors didn’t lock the model and code away on their servers. They made it a publicly available web application. This allows a bench scientist to search for interesting leads among billions of possible pairings with just a few clicks. It greatly lowers the barrier to using computational tools and is a good way to connect computation with experiments.
Of course, a nice interface doesn’t guarantee the quality of the predictions. In their case study, they validated 1,355 high-confidence drug-phenotype predictions. This is good, but it’s a form of retrospective validation—using existing literature to confirm the model’s predictions. The true test for this tool is whether it can predict new connections that are not yet discovered but can be confirmed by future experiments. That’s what will determine if it can change the game.
BIND is a powerful data integration and prediction engine, a “hypothesis generator” that packages complex knowledge graph technology into a friendly interface. It won’t tell you what the next blockbuster drug is, but it might give you a treasure map marking spots that seem absurd but could hold gold. Whether we dig up gold or rocks depends on our own judgment and lab work.
📜Paper: https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06789-5
3. AI Agent on Duty: Single-Cell Analysis Without the Tedious Scripts
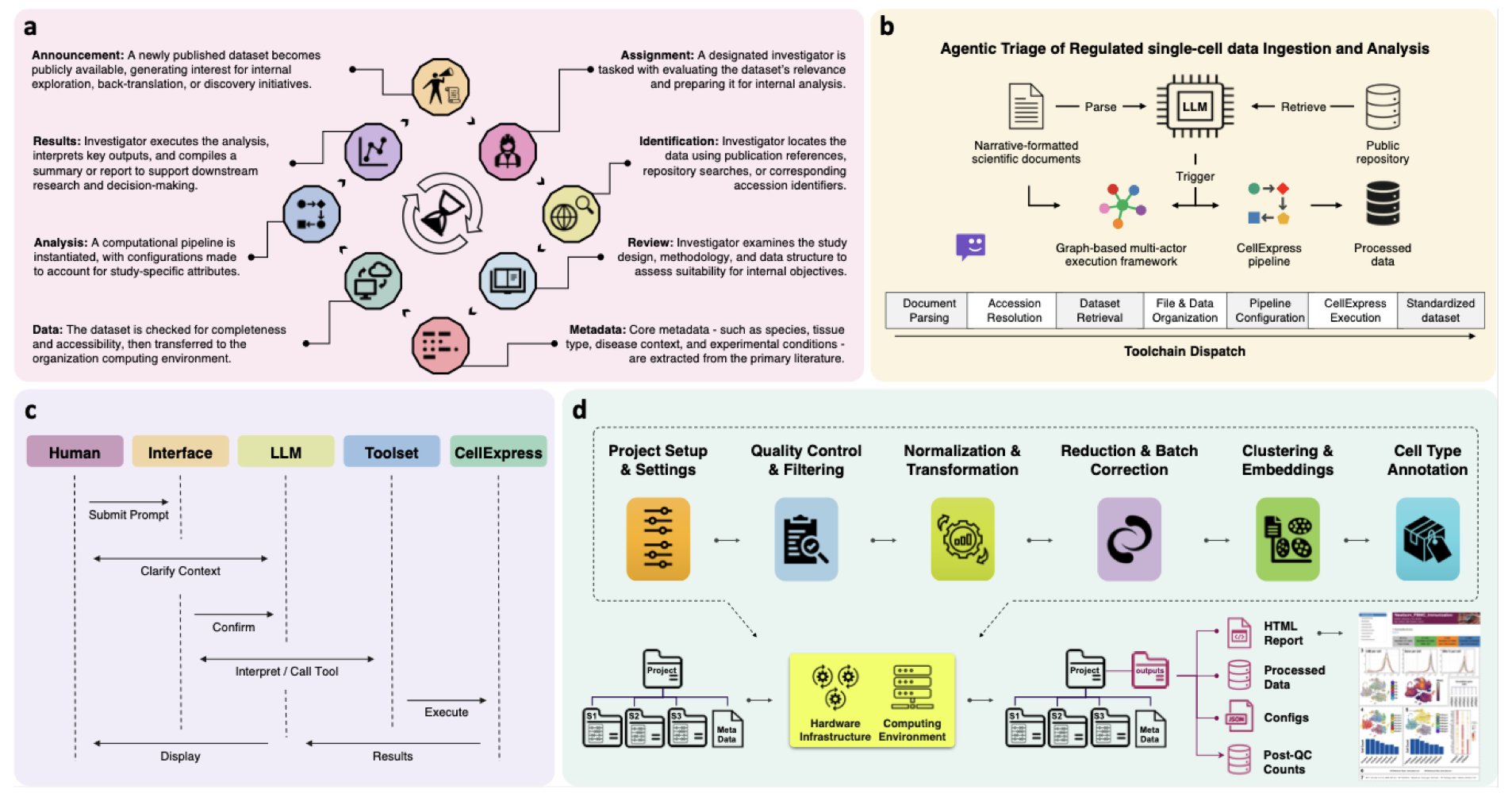
Anyone who has analyzed single-cell RNA-seq (scRNA-seq) data knows that the real challenge is often not the biological question, but the data preprocessing. Downloading data, organizing metadata, running the Seurat or Scanpy pipeline that always seems to have dependency errors… this tedious work can consume a large part of a PhD student’s life.
So, when a bioRxiv preprint mentioned an AI “agent” (Agentic AI) that could handle all this cumbersome work through conversation, it was both exciting and hard to believe. It sounds almost too good to be true, tailor-made for those who wrestle with data all day.
The tool is called CellAtria. Its main selling point isn’t just another automated script, but the “agent” concept itself.
What’s the difference? A traditional automated workflow is like a chef following a recipe exactly. If a step is missing or an ingredient is changed, it’s stuck. CellAtria’s architecture is more like a kitchen team. A “manager” (a Large Language Model, LLM) understands your order (“what you want to eat”) and then breaks the task down for different “specialists.” Some read papers, some find data in the GEO database, and others run the computational pipelines.
The entire workflow sounds futuristic. You can give it a paper and a one-sentence command, like “analyze the dataset on mouse lung fibroblasts from this paper.”
CellAtria then gets to work on its own. It parses the paper, finds the dataset accession number (like GSEXXXXX), automatically downloads it from the database, runs it through a built-in standardized pipeline called CellExpress, and gives you the results. You don’t have to write a single line of code.
The CellExpress pipeline itself is worth mentioning. It integrates the current mainstream scRNA-seq processing steps, ensuring standardization and reproducibility.
We all know that in bioinformatics, “reproducibility is the gold standard.” If everyone uses their own “secret sauce” to process data, it’s hard to compare results. CellAtria tries to solve this with a unified, high-quality pipeline, which is valuable in itself.
But the devil is in the details.
The system might work great for public datasets with perfect formatting and clear metadata. But what if a paper is vague, or a dataset’s metadata is a mess? Can the AI manager handle it? This is the key to it becoming a reliable research tool instead of just a neat toy.
The researchers seem to have considered transparency and control. The system has a real-time monitoring dashboard that lets users see what the AI is doing and intervene if needed.
The entire environment is packaged with Docker, which means you don’t have to go through the pain of setting up the environment. For anyone who has suffered from “dependency hell,” this is music to the ears.
CellAtria is more than just an efficiency tool. It’s an attempt at “research democratization.” It allows wet-lab scientists without a strong computational background to stand on the shoulders of giants and easily mine and reuse massive public datasets.
📜Title: An Agentic AI Framework for Ingestion and Standardization of Single-Cell RNA-seq Data Analysis 📜Paper: https://www.biorxiv.org/content/10.1101/2025.07.31.667880v1